

Exploiting Allosteric Properties of RAF and MEK Inhibitors to Target Therapy-Resistant Tumors Driven by Oncogenic BRAF Signaling
- PMID: 33568355
- PMCID: PMC8295204
- DOI: 10.1158/2159-8290.CD-20-1351
Exploiting Allosteric Properties of RAF and MEK Inhibitors to Target Therapy-Resistant Tumors Driven by Oncogenic BRAF Signaling
Abstract
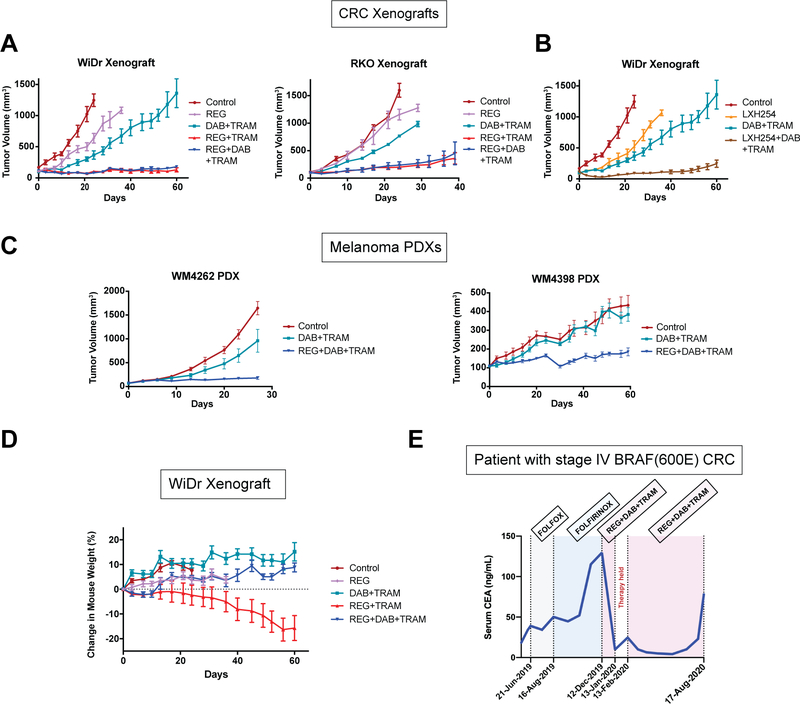
Current clinical RAF inhibitors (RAFi) inhibit monomeric BRAF (mBRAF) but are less potent against dimeric BRAF (dBRAF). RAFi equipotent for mBRAF and dBRAF have been developed but are predicted to have lower therapeutic index. Here we identify a third class of RAFi that selectively inhibits dBRAF over mBRAF. Molecular dynamic simulations reveal restriction of the movement of the BRAF αC-helix as the basis of inhibitor selectivity. Combination of inhibitors based on their conformation selectivity (mBRAF- plus dBRAF-selective plus the most potent BRAF-MEK disruptor MEK inhibitor) promoted suppression of tumor growth in BRAFV600E therapy-resistant models. Strikingly, the triple combination showed no toxicities, whereas dBRAF-selective plus MEK inhibitor treatment caused weight loss in mice. Finally, the triple combination achieved durable response and improved clinical well-being in a patient with stage IV colorectal cancer. Thus, exploiting allosteric properties of RAF and MEK inhibitors enables the design of effective and well-tolerated therapies for BRAFV600E tumors. SIGNIFICANCE: This work identifies a new class of RAFi that are selective for dBRAF over mBRAF and determines the basis of their selectivity. A rationally designed combination of RAF and MEK inhibitors based on their conformation selectivity achieved increased efficacy and a high therapeutic index when used to target BRAFV600E tumors.See related commentary by Zhang and Bollag, p. 1620.This article is highlighted in the In This Issue feature, p. 1601.
©2021 American Association for Cancer Research.
Conflict of interest statement
No competing financial interests.
Figures
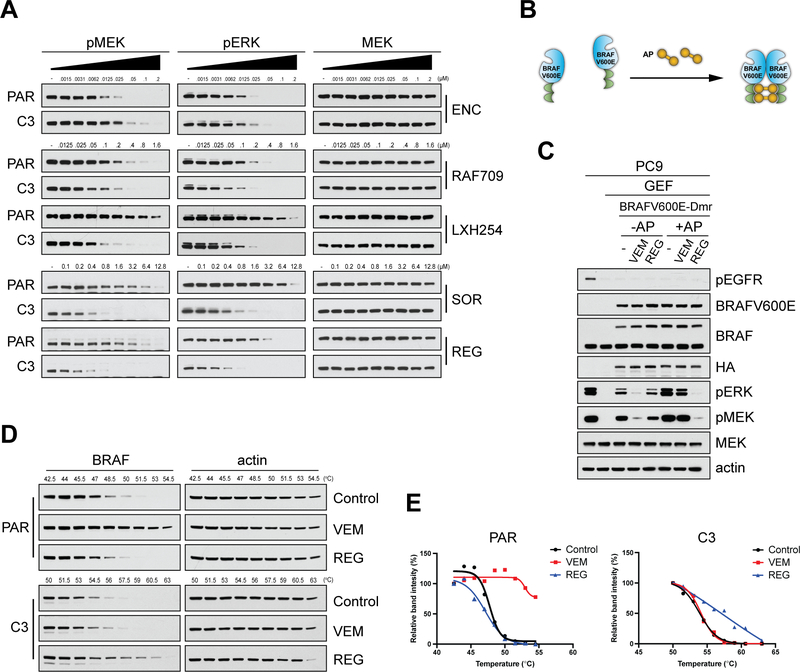
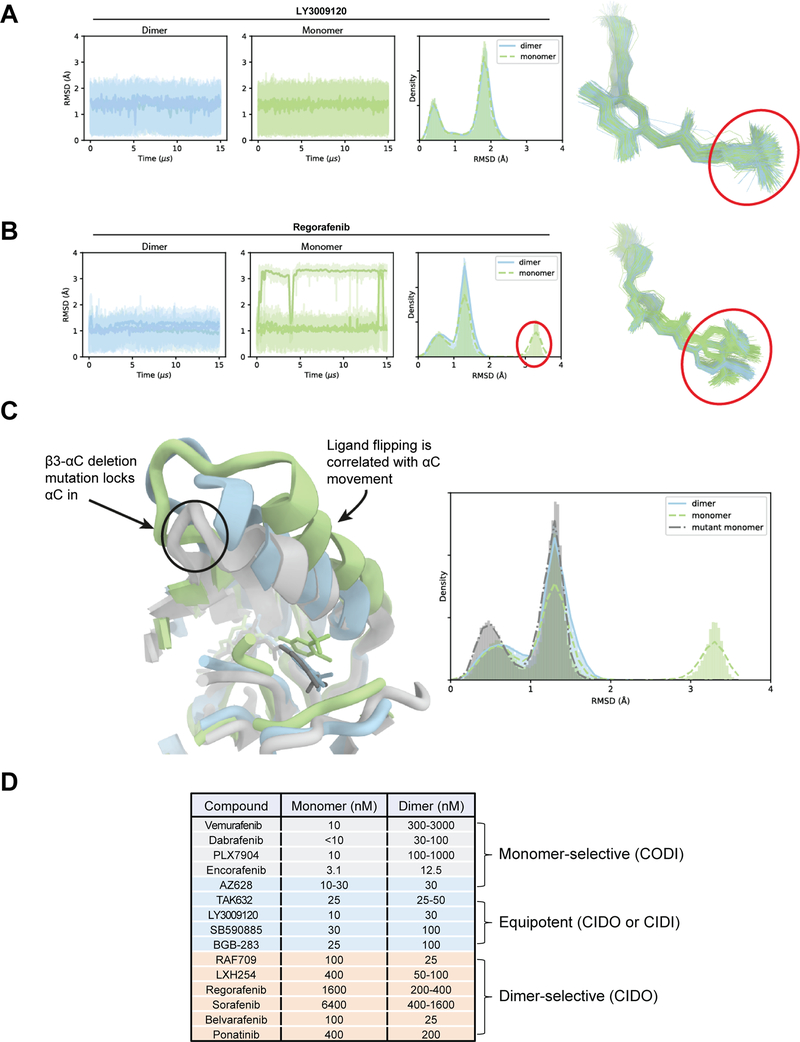

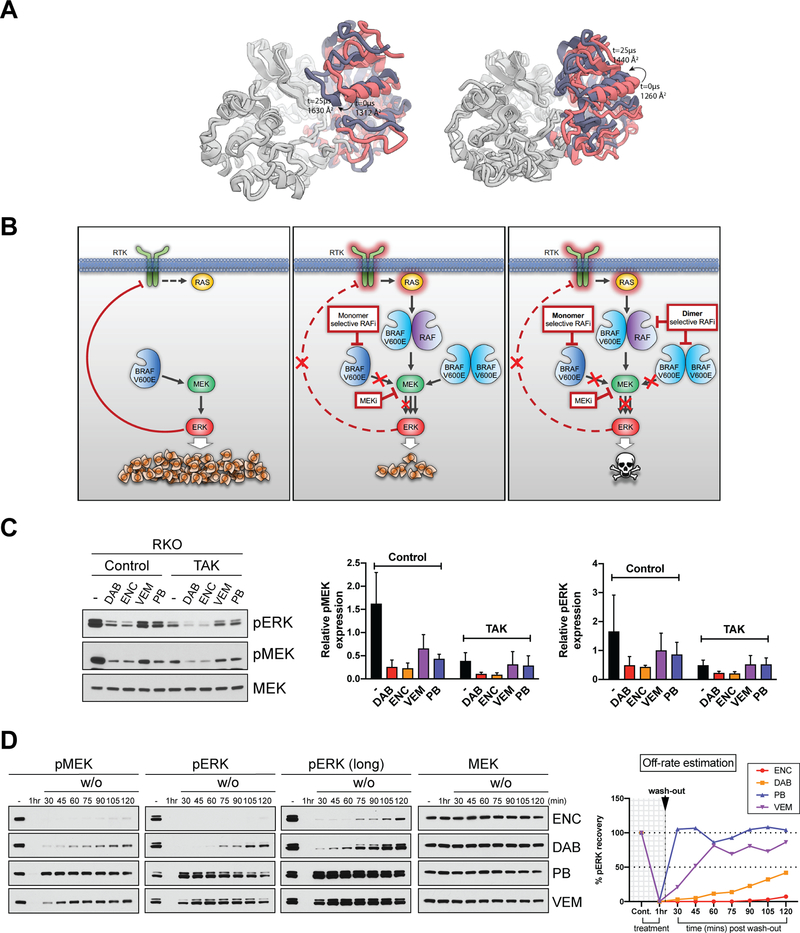

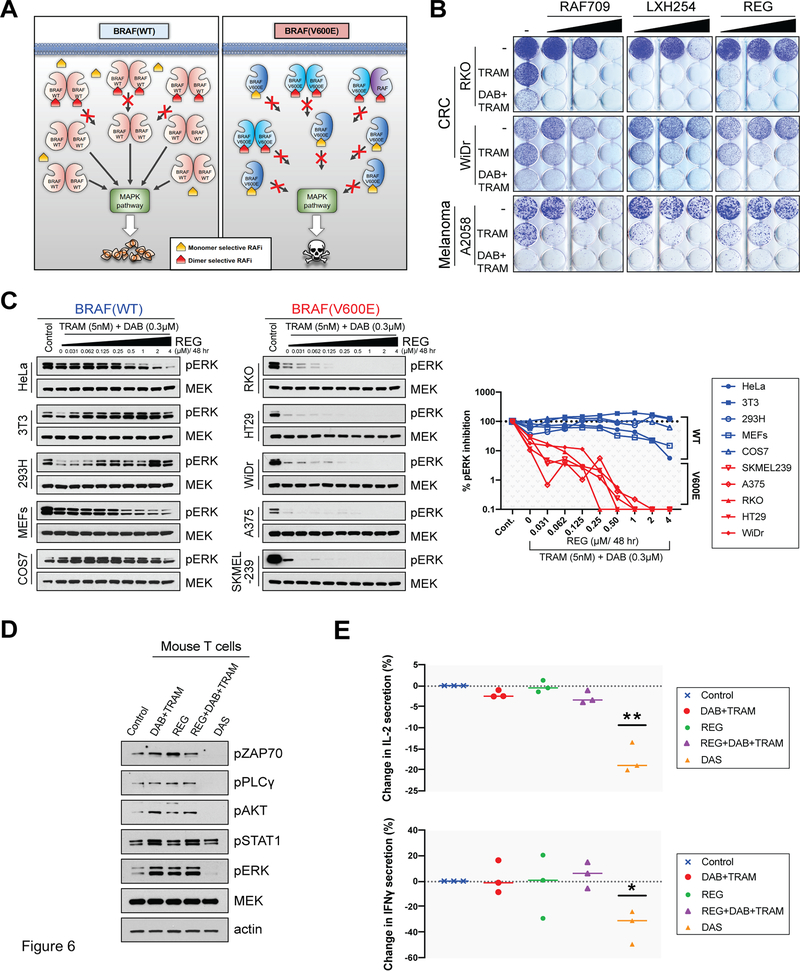
Comment in
-
Triple Therapy to Outwit the BRAF Oncogene.Cancer Discov. 2021 Jul;11(7):1620-1622. doi: 10.1158/2159-8290.CD-21-0378. Cancer Discov. 2021. PMID: 34284995
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
