

Human neurons from Christianson syndrome iPSCs reveal mutation-specific responses to rescue strategies
- PMID: 33568516
- PMCID: PMC7888381
- DOI: 10.1126/scitranslmed.aaw0682
Human neurons from Christianson syndrome iPSCs reveal mutation-specific responses to rescue strategies
Abstract
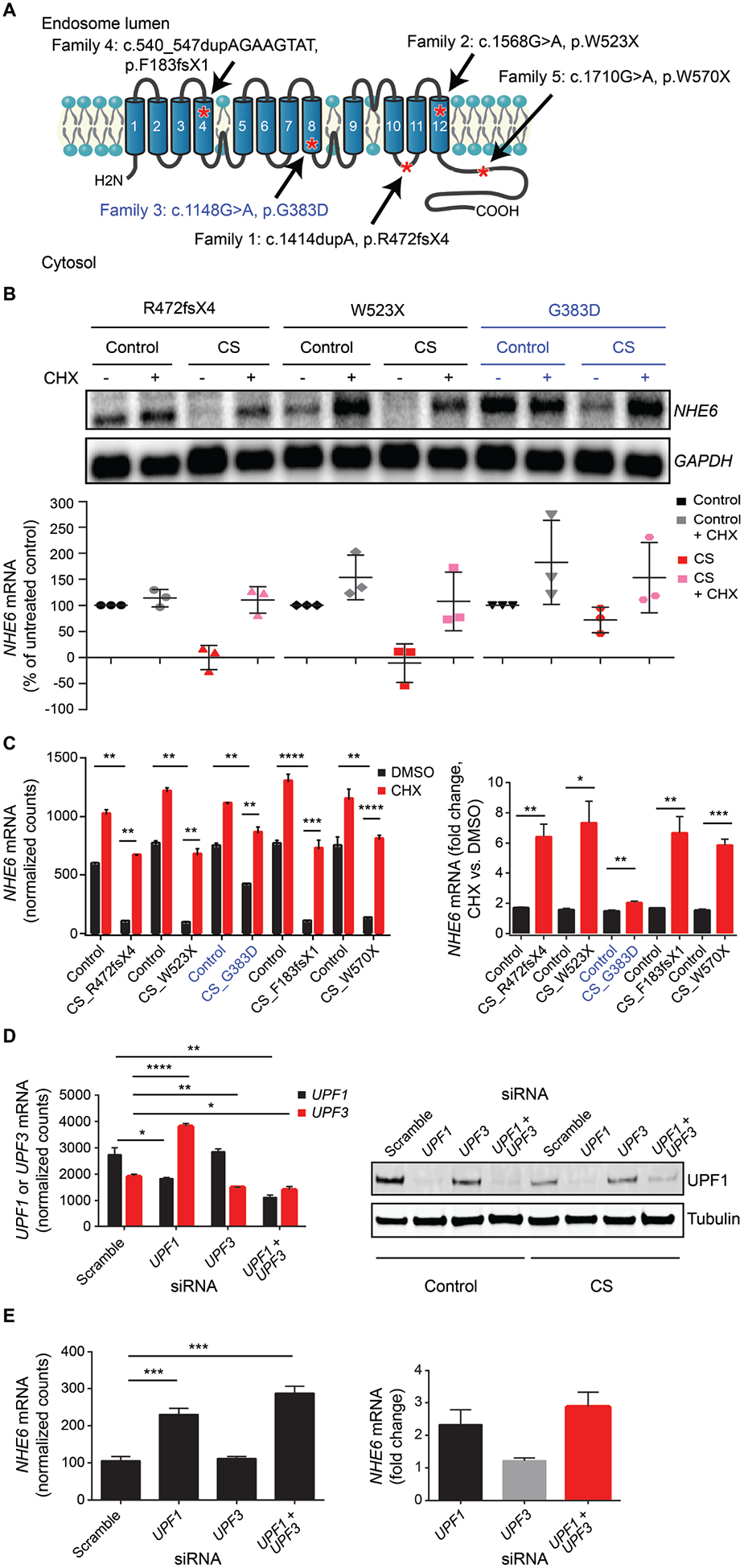
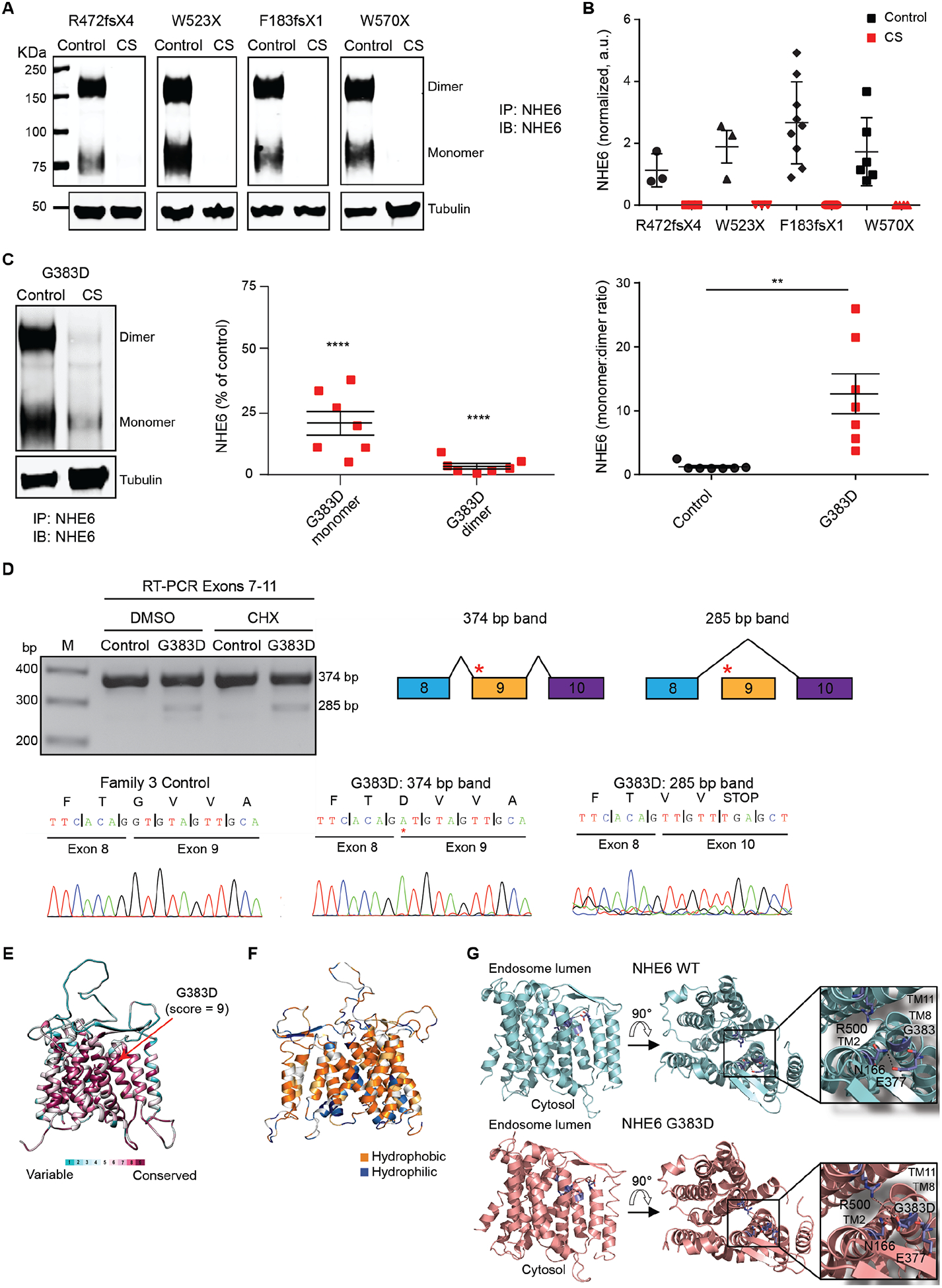
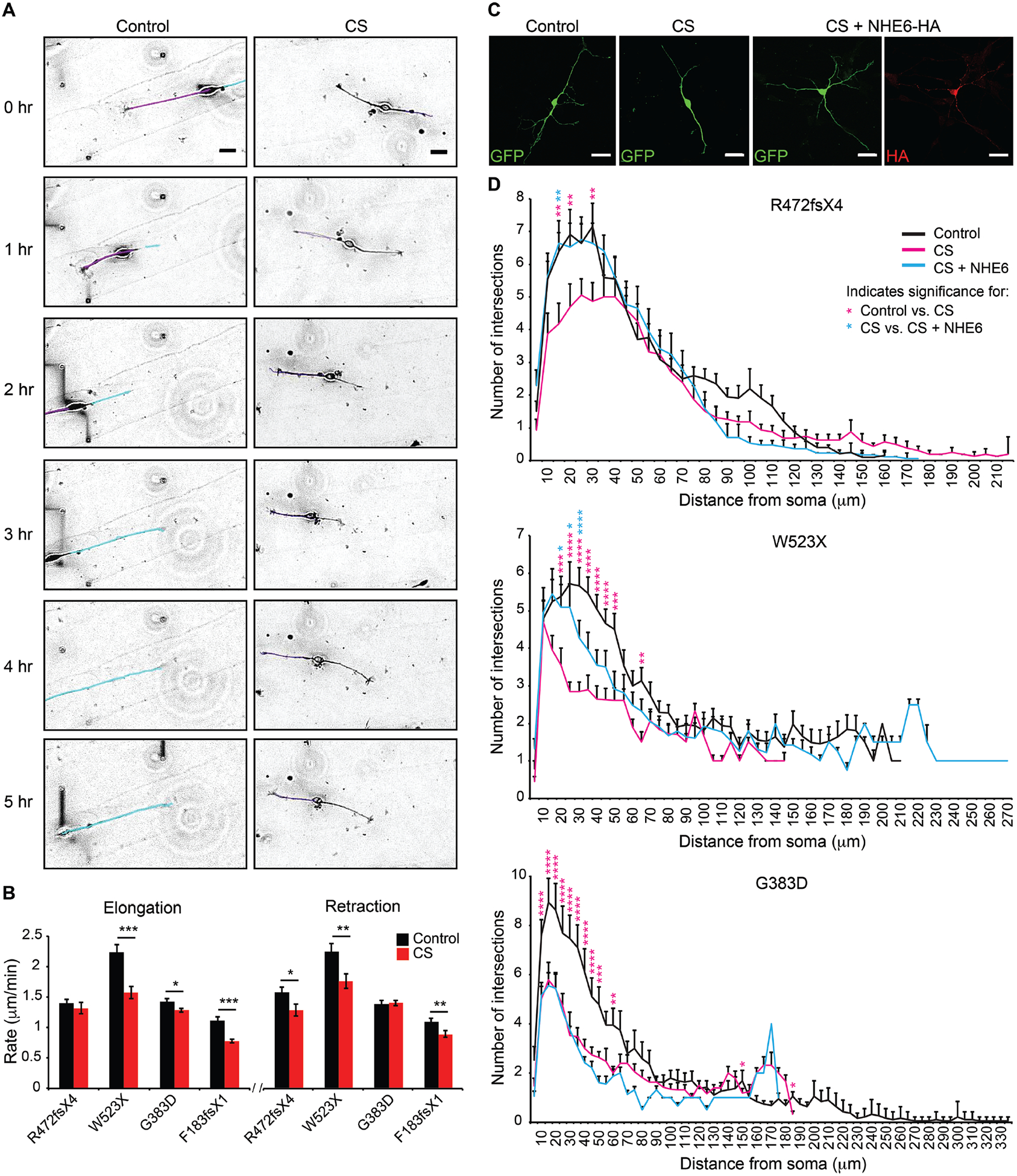
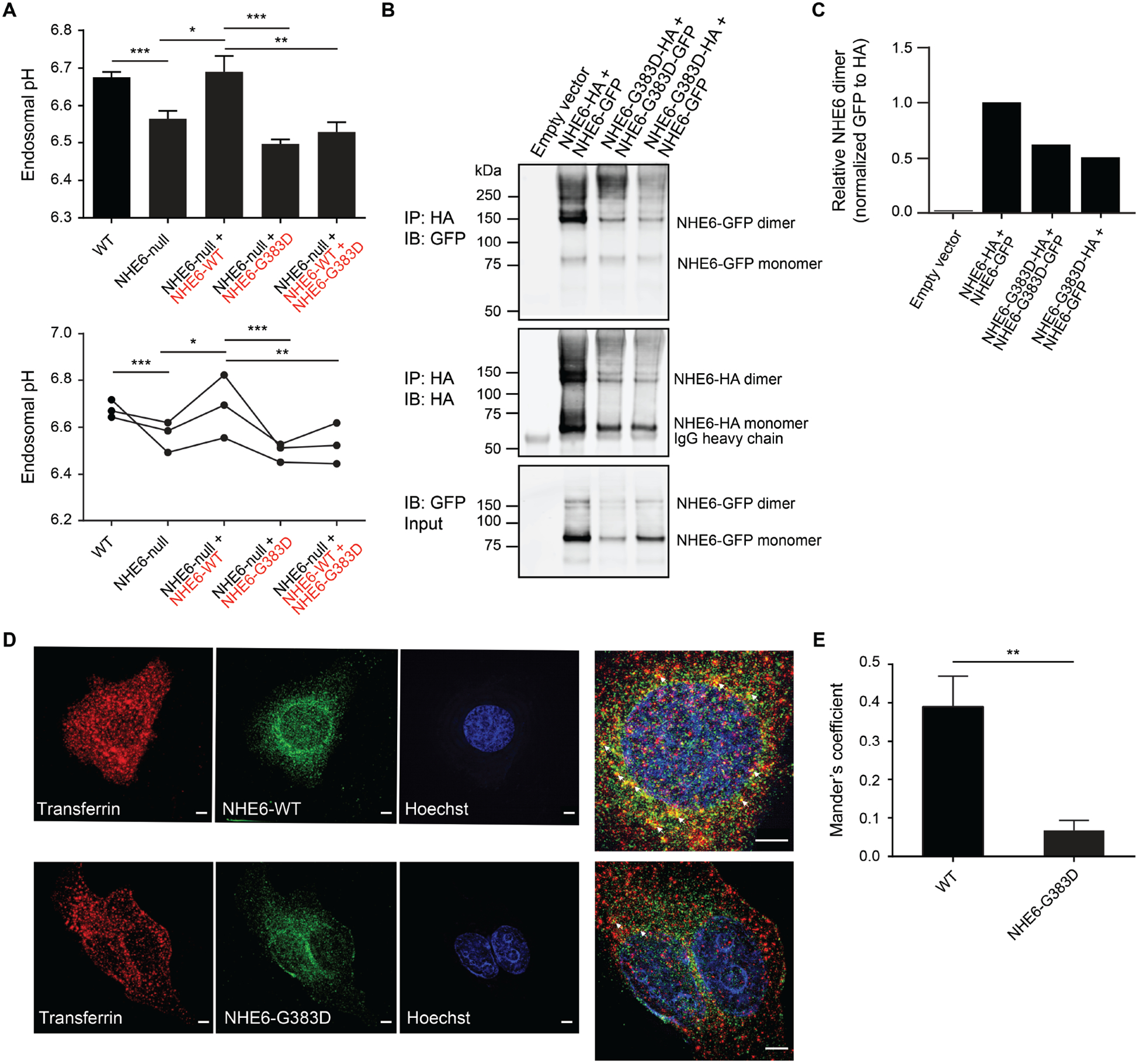
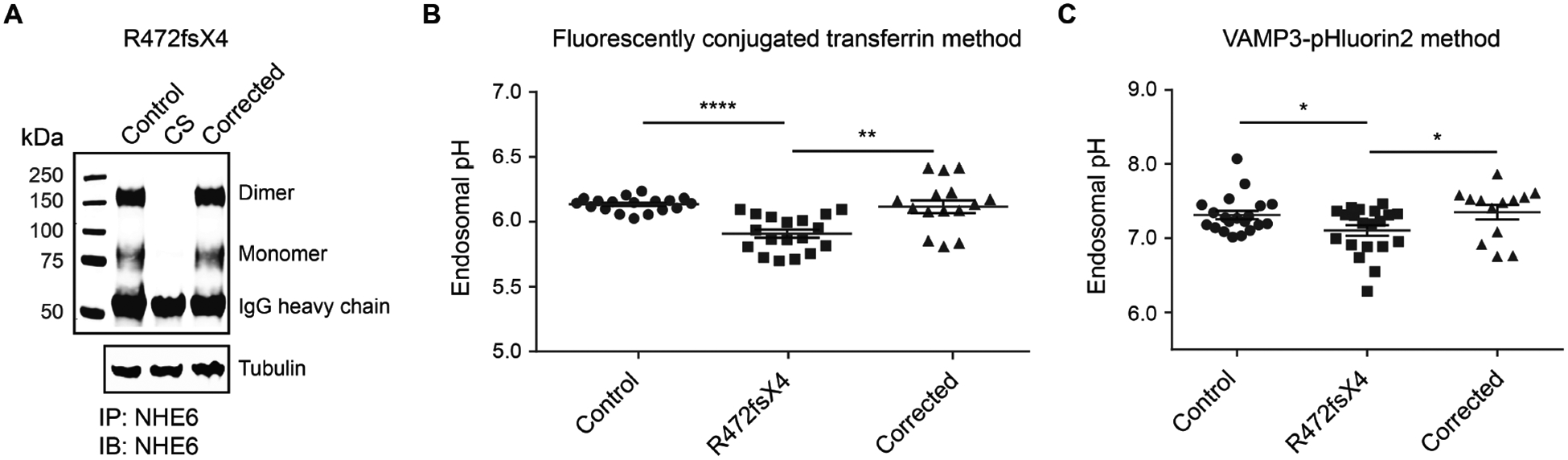
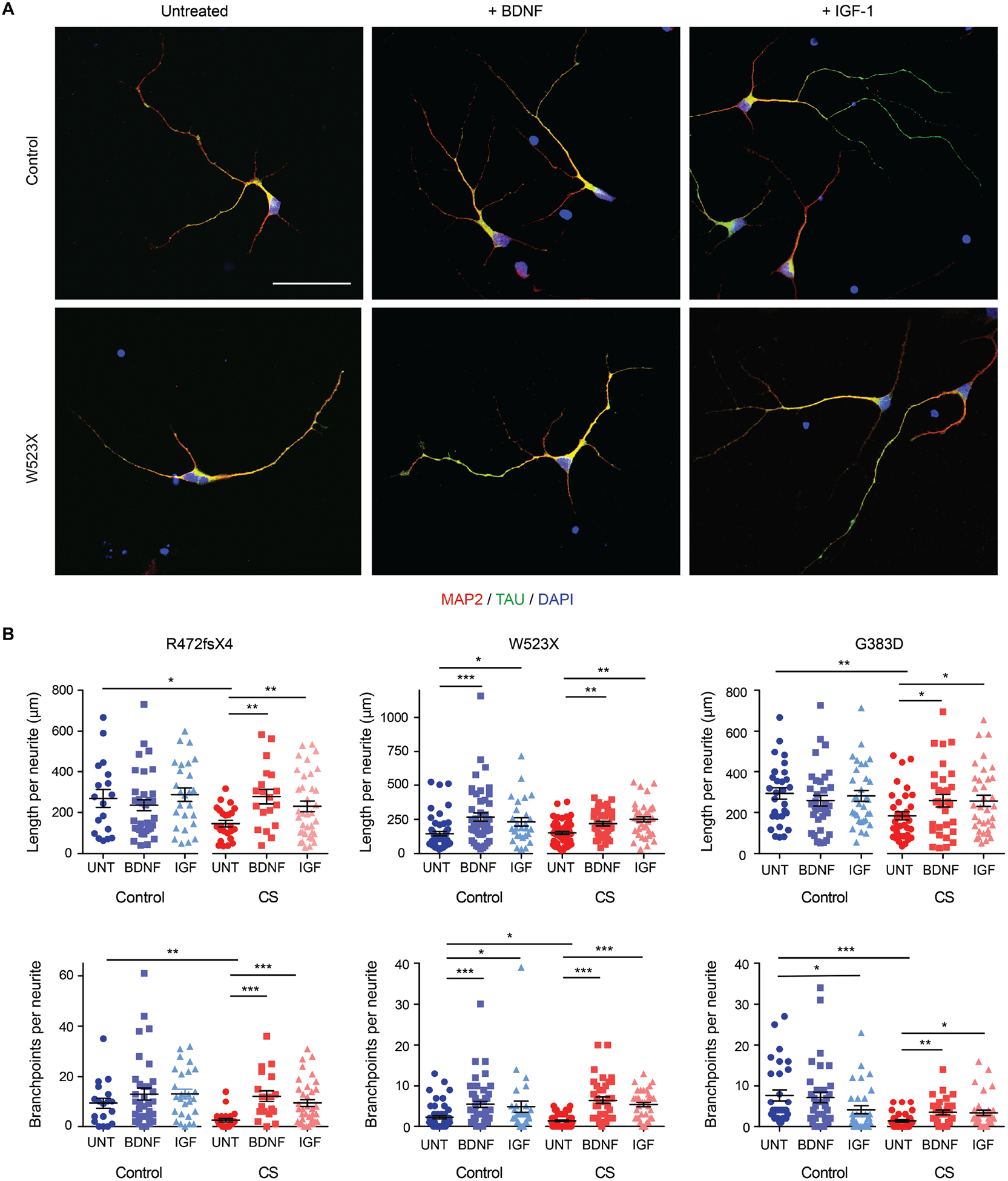
Christianson syndrome (CS), an X-linked neurological disorder characterized by postnatal attenuation of brain growth (postnatal microcephaly), is caused by mutations in SLC9A6, the gene encoding endosomal Na+/H+ exchanger 6 (NHE6). To hasten treatment development, we established induced pluripotent stem cell (iPSC) lines from patients with CS representing a mutational spectrum, as well as biologically related and isogenic control lines. We demonstrated that pathogenic mutations lead to loss of protein function by a variety of mechanisms: The majority of mutations caused loss of mRNA due to nonsense-mediated mRNA decay; however, a recurrent, missense mutation (the G383D mutation) had both loss-of-function and dominant-negative activities. Regardless of mutation, all patient-derived neurons demonstrated reduced neurite growth and arborization, likely underlying diminished postnatal brain growth in patients. Phenotype rescue strategies showed mutation-specific responses: A gene transfer strategy was effective in nonsense mutations, but not in the G383D mutation, wherein residual protein appeared to interfere with rescue. In contrast, application of exogenous trophic factors (BDNF or IGF-1) rescued arborization phenotypes across all mutations. These results may guide treatment development in CS, including gene therapy strategies wherein our data suggest that response to treatment may be dictated by the class of mutation.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Figures
References
-
- Christianson AL, Stevenson RE, van der Meyden CH, Pelser J, Theron FW, van Rensburg PL, Chandler M, Schwartz CE, X linked severe mental retardation, craniofacial dysmorphology, epilepsy, ophthalmoplegia, and cerebellar atrophy in a large South African kindred is localised to Xq24-q27. J Med Genet 36, 759–766 (1999). - PMC - PubMed
-
- Gilfillan GD, Selmer KK, Roxrud I, Smith R, Kyllerman M, Eiklid K, Kroken M, Mattingsdal M, Egeland T, Stenmark H, Sjoholm H, Server A, Samuelsson L, Christianson A, Tarpey P, Whibley A, Stratton MR, Futreal PA, Teague J, Edkins S, Gecz J, Turner G, Raymond FL, Schwartz C, Stevenson RE, Undlien DE, Stromme P, SLC9A6 mutations cause X-linked mental retardation, microcephaly, epilepsy, and ataxia, a phenotype mimicking Angelman syndrome. Am J Hum Genet 82, 1003–1010 (2008). - PMC - PubMed
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
