

The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling
- PMID: 33571454
- PMCID: PMC8035082
- DOI: 10.1016/j.cmet.2021.01.015
The glucose-dependent insulinotropic polypeptide (GIP) regulates body weight and food intake via CNS-GIPR signaling
Abstract
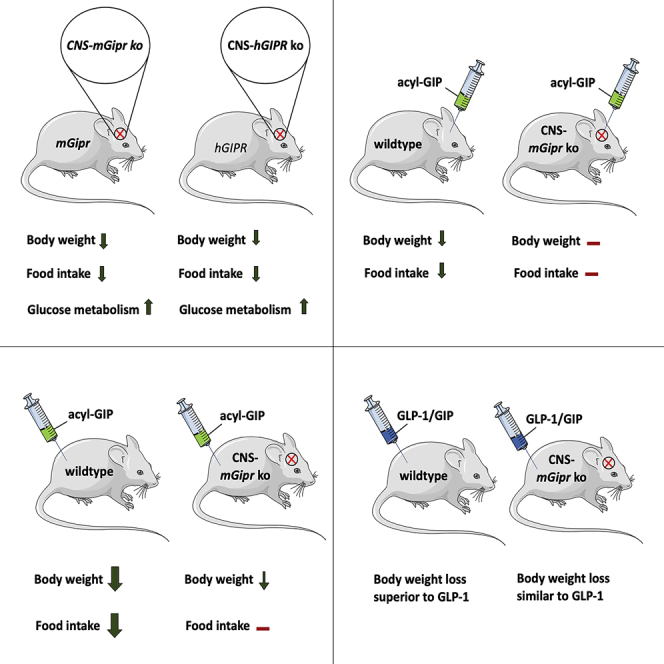
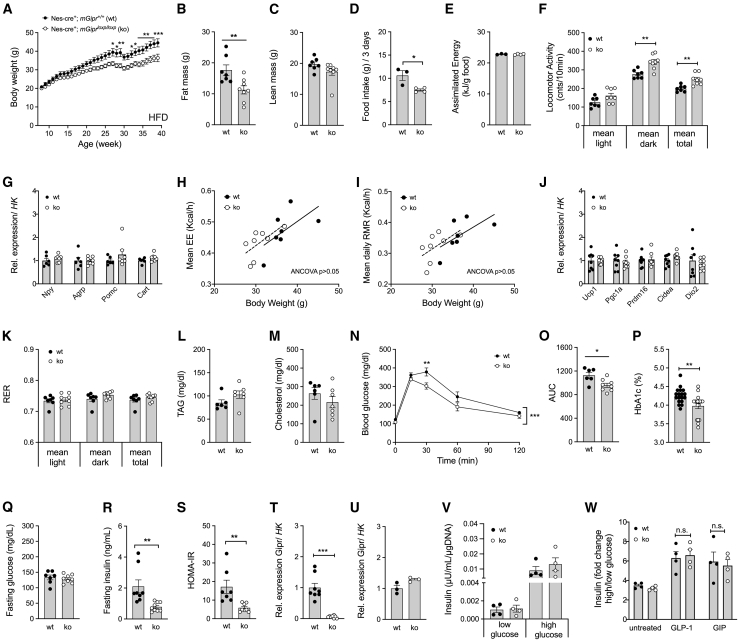
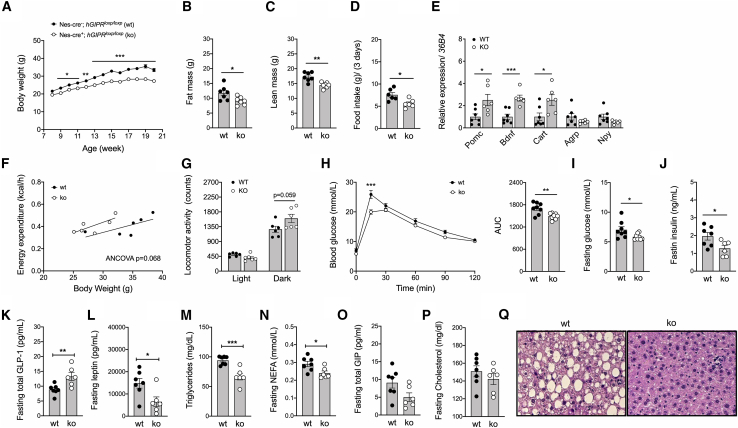
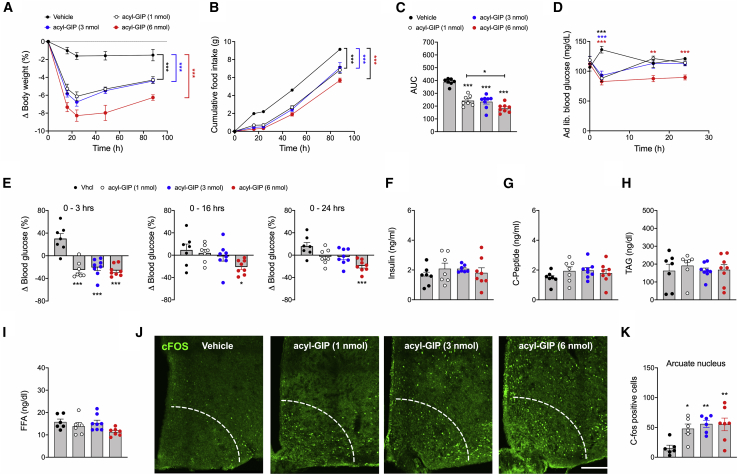
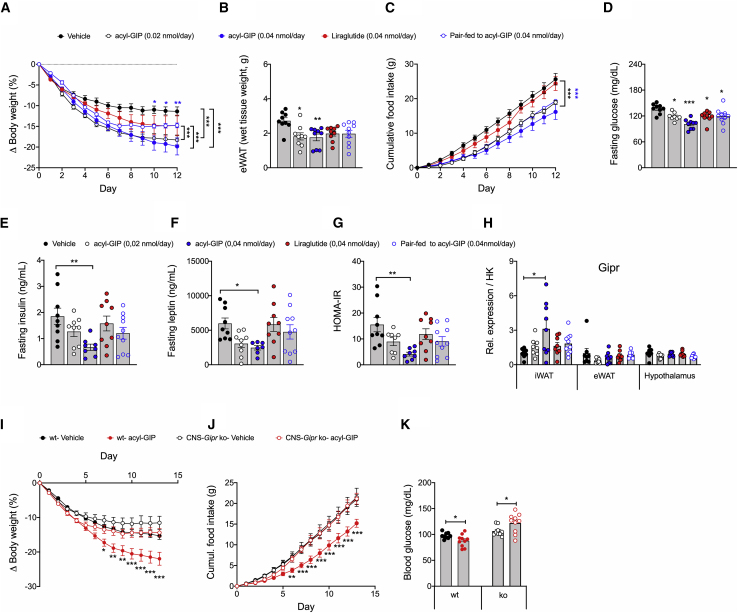
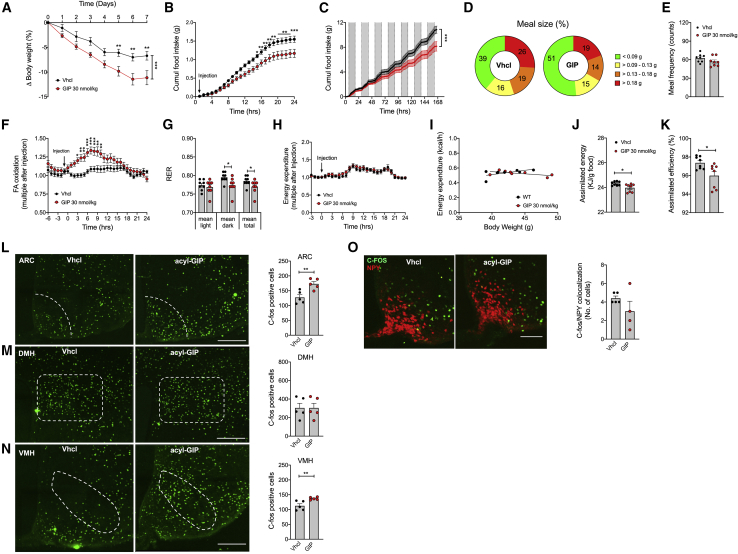
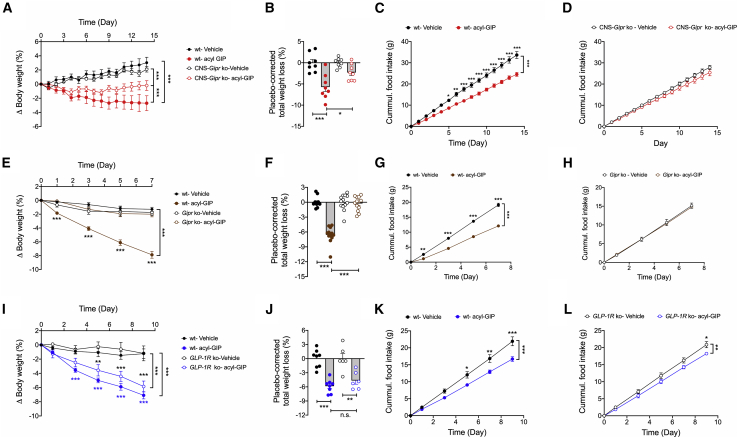
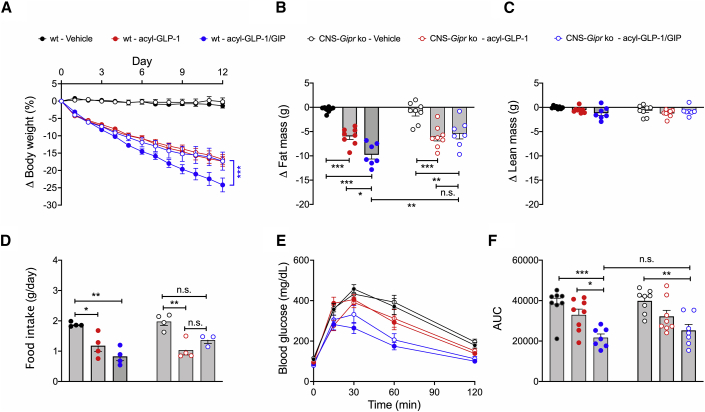
Uncertainty exists as to whether the glucose-dependent insulinotropic polypeptide receptor (GIPR) should be activated or inhibited for the treatment of obesity. Gipr was recently demonstrated in hypothalamic feeding centers, but the physiological relevance of CNS Gipr remains unknown. Here we show that HFD-fed CNS-Gipr KO mice and humanized (h)GIPR knockin mice with CNS-hGIPR deletion show decreased body weight and improved glucose metabolism. In DIO mice, acute central and peripheral administration of acyl-GIP increases cFos neuronal activity in hypothalamic feeding centers, and this coincides with decreased body weight and food intake and improved glucose handling. Chronic central and peripheral administration of acyl-GIP lowers body weight and food intake in wild-type mice, but shows blunted/absent efficacy in CNS-Gipr KO mice. Also, the superior metabolic effect of GLP-1/GIP co-agonism relative to GLP-1 is extinguished in CNS-Gipr KO mice. Our data hence establish a key role of CNS Gipr for control of energy metabolism.
Keywords: CNS; GIP; GIPR CNS KO; body weight; diet-induced obesity; food intake; glucose metabolism; incretin; type 2 diabetes.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests M.H.T. is a member of the scientific advisory board of ERX Pharmaceuticals, Cambridge, MA. He was a member of the Research Cluster Advisory Panel (ReCAP) of the Novo Nordisk Foundation between 2017 and 2019. He attended a scientific advisory board meeting of the Novo Nordisk Foundation Center for Basic Metabolic Research, University of Copenhagen, in 2016. He received funding for his research projects from Novo Nordisk (2016–2020) and Sanofi-Aventis (2012–2019). He was a consultant for Bionorica SE (2013–2017), Menarini Ricerche S.p.A. (2016), and Bayer Pharma AG Berlin (2016). As former Director of the Helmholtz Diabetes Center and the Institute for Diabetes and Obesity at Helmholtz Zentrum München (2011–2018), and since 2018, as CEO of Helmholtz Zentrum München, he has been responsible for collaborations with a multitude of companies and institutions worldwide. In this capacity, he discussed potential projects with and has signed/signs contracts for his institute(s) and for the staff for research funding and/or collaborations with industry and academia worldwide, including, but not limited to, pharmaceutical corporations like Boehringer Ingelheim, Eli Lilly, Novo Nordisk, Medigene, Arbormed, BioSyngen, and others. In this role, he was/is further responsible for commercial technology transfer activities of his institute(s), including diabetes-related patent portfolios of Helmholtz Zentrum München as, e.g., WO/2016/188932 A2 or WO/2017/194499 A1. M.H.T. confirms that to the best of his knowledge none of the above funding sources were involved in the preparation of this paper. T.D.M. and K.S. receive research funding from Novo Nordisk, but these funds are unrelated to the here described work. D.J.D. has received speaking and consulting fees from Merck and Novo Nordisk Inc. and consulting fees from Forkhead Biopharmaceuticals and Kallyope Inc. R.D.D. is a co-inventor on intellectual property owned by Indiana University and licensed to Novo Nordisk. He was previously employed by Novo Nordisk. P.J.K., S.A.M., and B.F. are current employees of Novo Nordisk.
Figures
References
-
- Asmar M., Asmar A., Simonsen L., Gasbjerg L.S., Sparre-Ulrich A.H., Rosenkilde M.M., Hartmann B., Dela F., Holst J.J., Bülow J. The gluco- and liporegulatory and vasodilatory effects of glucose-dependent insulinotropic polypeptide (GIP) are abolished by an antagonist of the human GIP receptor. Diabetes. 2017;66:2363–2371. - PubMed
-
- Bastidas-Ponce A., Tritschler S., Dony L., Scheibner K., Tarquis-Medina M., Salinno C., Schirge S., Burtscher I., Böttcher A., Theis F.J. Comprehensive single cell mRNA profiling reveals a detailed roadmap for pancreatic endocrinogenesis. Development. 2019;146:dev173849. - PubMed
-
- Beck B., Max J.P. Direct metabolic effects of gastric inhibitory polypeptide (GIP): dissociation at physiological levels of effects on insulin-stimulated fatty acid and glucose incorporation in rat adipose tissue. Diabetologia. 1986;29:68. - PubMed
-
- Campbell J.E., Ussher J.R., Mulvihill E.E., Kolic J., Baggio L.L., Cao X., Liu Y., Lamont B.J., Morii T., Streutker C.J. TCF1 links GIPR signaling to the control of beta cell function and survival. Nat. Med. 2016;22:84–90. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
