

SPG9A with the new occurrence of an ALDH18A1 mutation in a CMT1A family with PMP22 duplication: case report
- PMID: 33573605
- PMCID: PMC7876803
- DOI: 10.1186/s12883-021-02087-x
SPG9A with the new occurrence of an ALDH18A1 mutation in a CMT1A family with PMP22 duplication: case report
Abstract
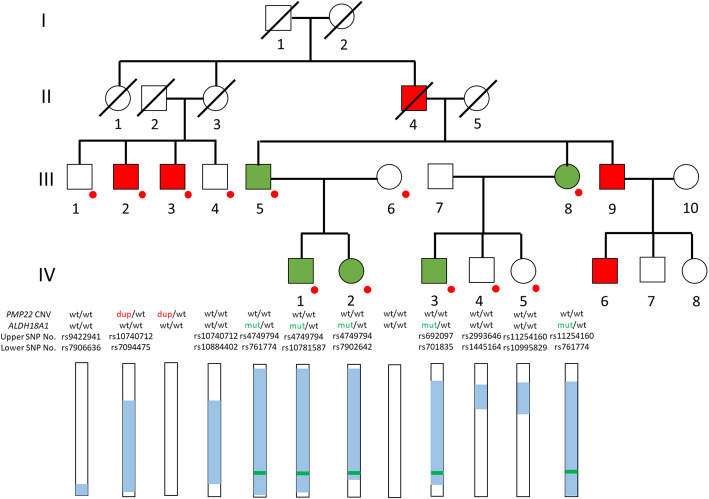
Background: ALDH18A1 mutations lead to delta-1-pyrroline-5-carboxylate-synthetase (P5CS) deficiency, which is a urea cycle-related disorder including SPG9A, SPG9B, autosomal dominant cutis laxa-3 (ADCL3), and autosomal recessive cutis laxa type 3A (ARCL3A). These diseases exhibit a broad clinical spectrum, which makes the diagnosis of P5CS deficiency difficult. We report here a rare Japanese family including both patients with an ALDH18A1 mutation (SPG9A) and ones with CMT1A.
Case presentation: A Japanese family included five patients with the CMT phenotype and five with the HSP phenotype in four generations. The patients with the HSP phenotype showed a pure or complicated form, and intrafamilial clinical variability was noted. Genetically, FISH analysis revealed that two CMT patients had a PMP22 duplication (CMT1A). Exome analysis and Sanger sequencing revealed five HSP patients had an ALDH18A1 heterozygous mutation of c.755G > A, which led to SPG9A. Haplotype analysis revealed that the ALDH18A1 mutation must have newly occurred. To date, although de novo mutations of ALDH18A1 have been described in ADCL3A, they were not mentioned in SPG9A in earlier reports. Thus, this is the first SPG9A family with a de novo mutation or the new occurrence of gonadal mosaicism of ALDH18A1. Analysis of serum amino acid levels revealed that two SPG9A patients and two unaffected family members had low citrulline levels and one had a low level of ornithine.
Conclusions: Since the newly occurring ALDH18A1 mutation, c.755G > A, is the same as that in two ADHSP families and one sporadic patient with SPG9A reported previously, this genomic site might easily undergo mutation. The patients with the c.755G > A mutation in our family showed clinical variability of symptoms like in the earlier reported two families and one sporadic patient with this mutation. Further studies are required to clarify the relationship between the amino acid levels and clinical manifestations, which will reveal how P5CS deficiency influences disease phenotypes including ARCL3A, ADCL3, SPG9B, and SPG9A.
Keywords: ALDH18A1; Charcot-Marie-tooth disease; Gonadal mosaicism; PMP22; SPG9A; de novo mutation.
Conflict of interest statement
The authors disclose no potential conflicts of interest.
Figures
References
-
- Coutelier M, Goizet C, Durr A, Habarou F, Morais S, Dionne-Laporte A, Tao F, Konop J, Stoll M, Charles P, Jacoupy M, Matusiak R, Alonso I, Tallaksen C, Mairey M, Kennerson M, Gaussen M, Schule R, Janin M, Morice-Picard F, Durand CM, Depienne C, Calvas P, Coutinho P, Saudubray JM, Rouleau G, Brice A, Nicholson G, Dariou F, Loureiro JL, Zuchner S, Ottolenghi C, Mochel F, Stevanin G. Alteration of ornithine metabolism leads to dominant and recessive hereditary spastic paraplegia. Brain. 2015;138:2191–2205. doi: 10.1093/brain/awv143. - DOI - PMC - PubMed
-
- Koh K, Ishiura H, Beppu M, Shimazaki H, Ichinose Y, Mitsui J, Kuwabara S, Tsuji S, Takiyama Y, Japan Spastic Paraplegia Research Consortium. Novel mutations in the ALDH18A1 gene in complicated hereditary spastic paraplegia with cerebellar ataxia and cognitive impairment. J Hum Genet. 2018;63:1009–13. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
