

Liquid chromatin Hi-C characterizes compartment-dependent chromatin interaction dynamics
- PMID: 33574602
- PMCID: PMC7946813
- DOI: 10.1038/s41588-021-00784-4
Liquid chromatin Hi-C characterizes compartment-dependent chromatin interaction dynamics
Abstract
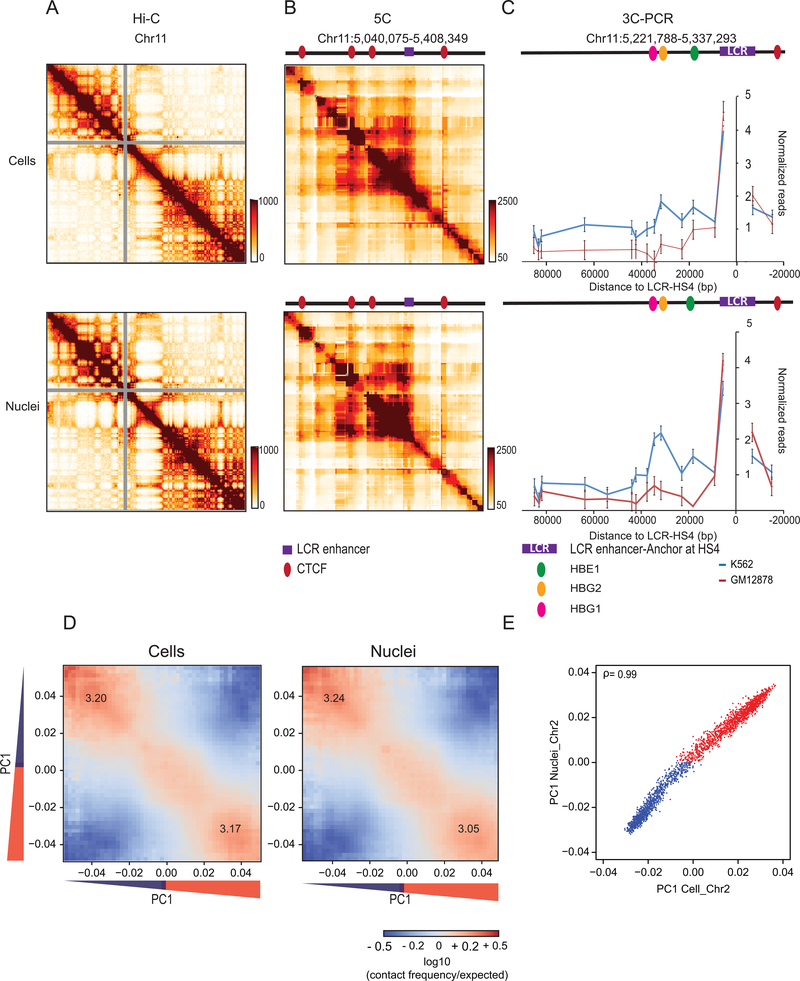
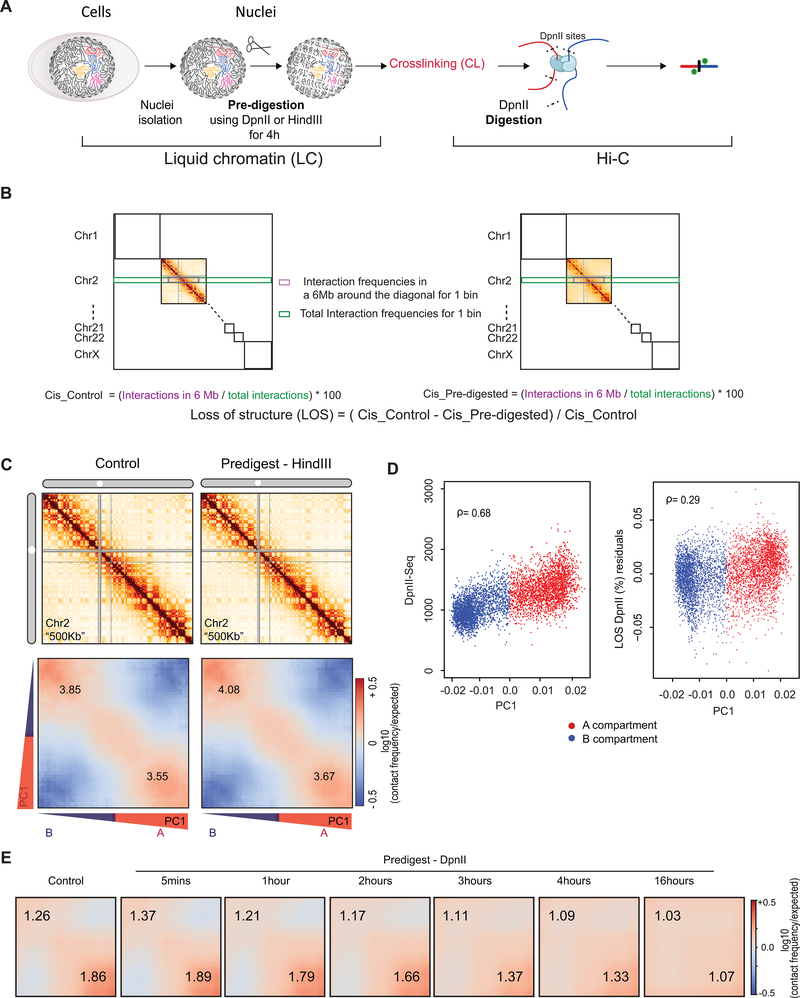
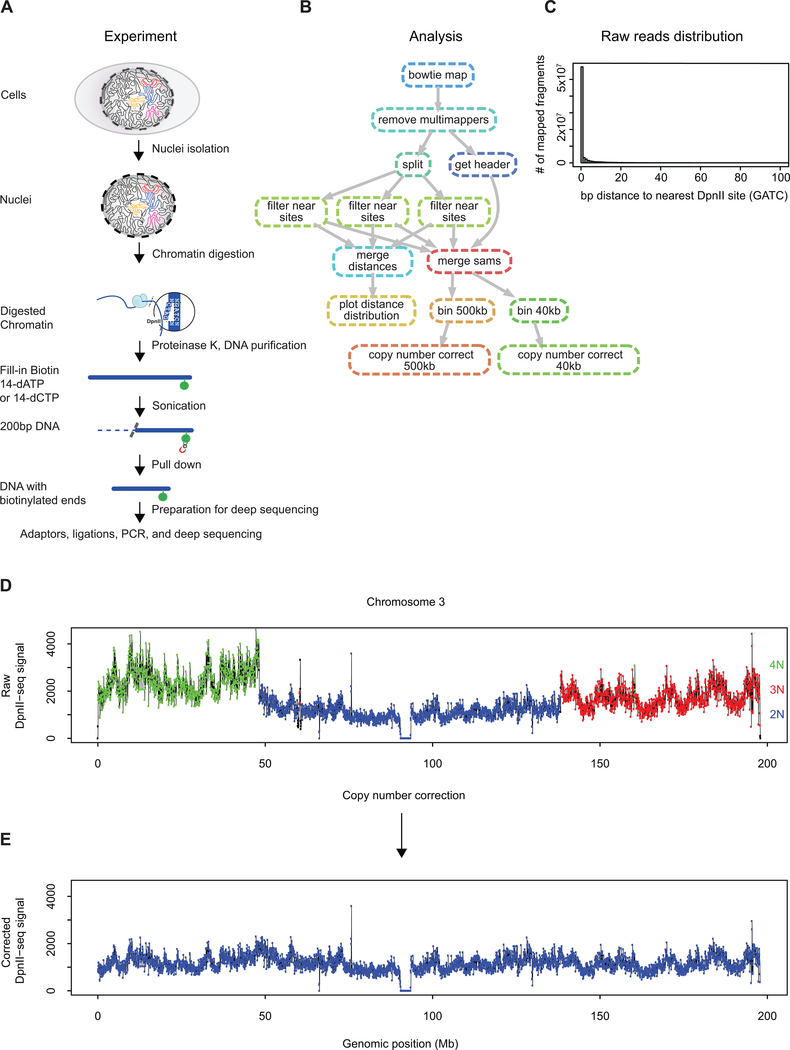
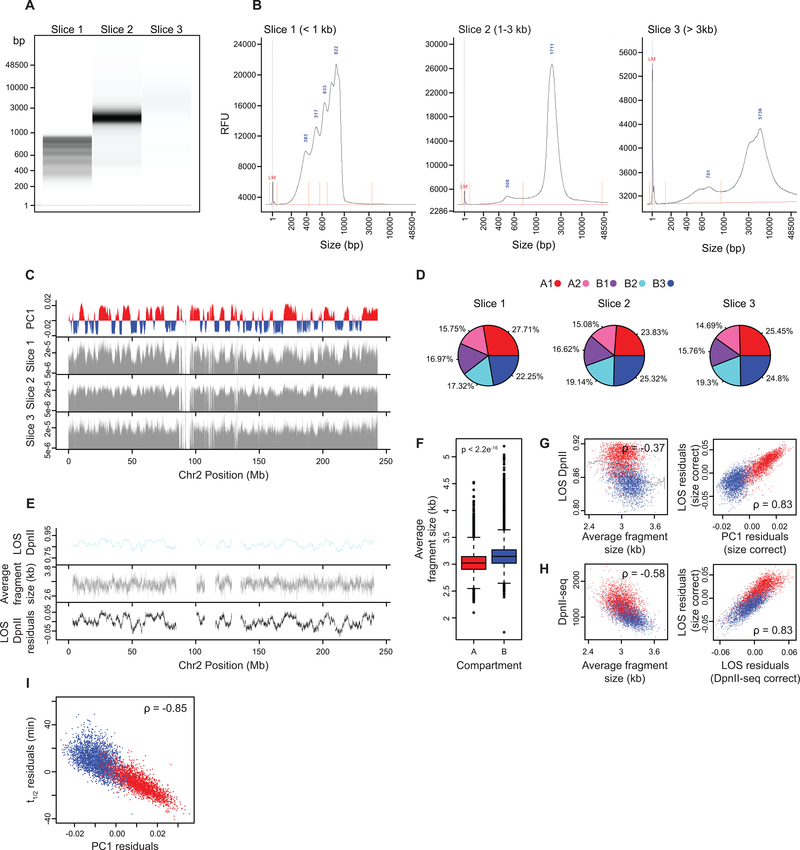
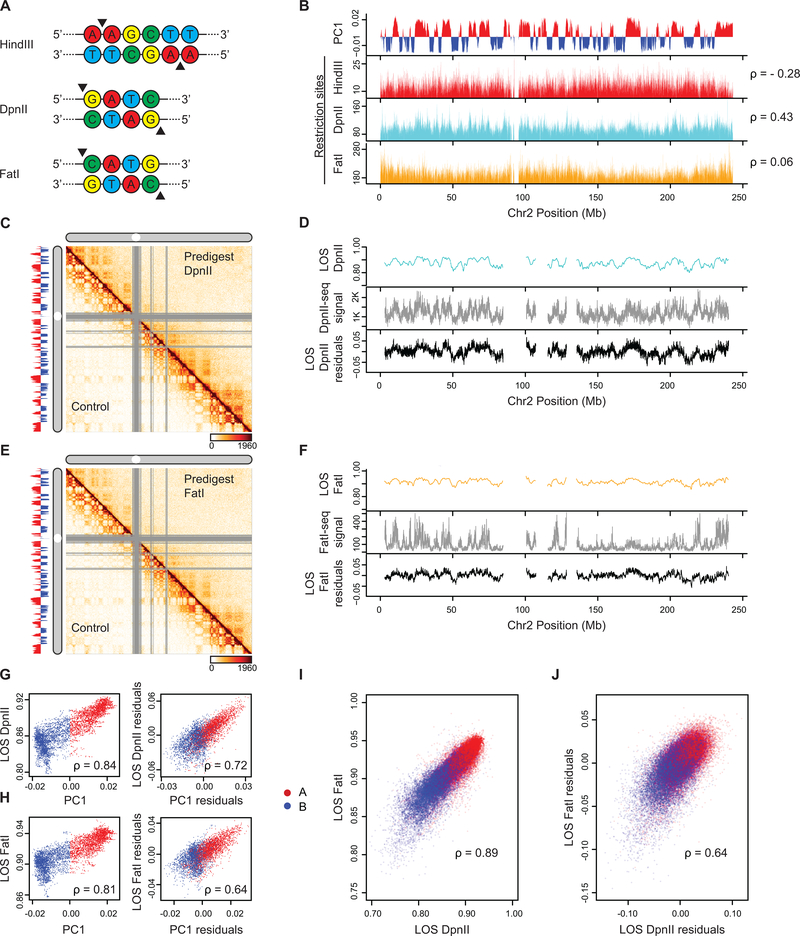
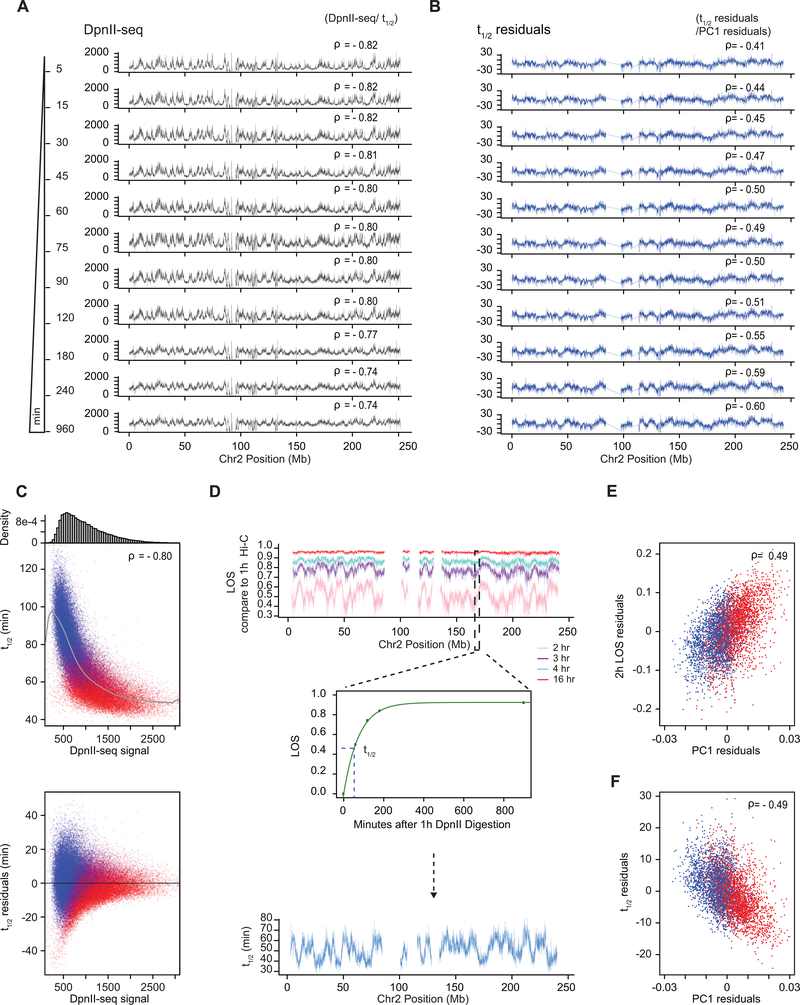
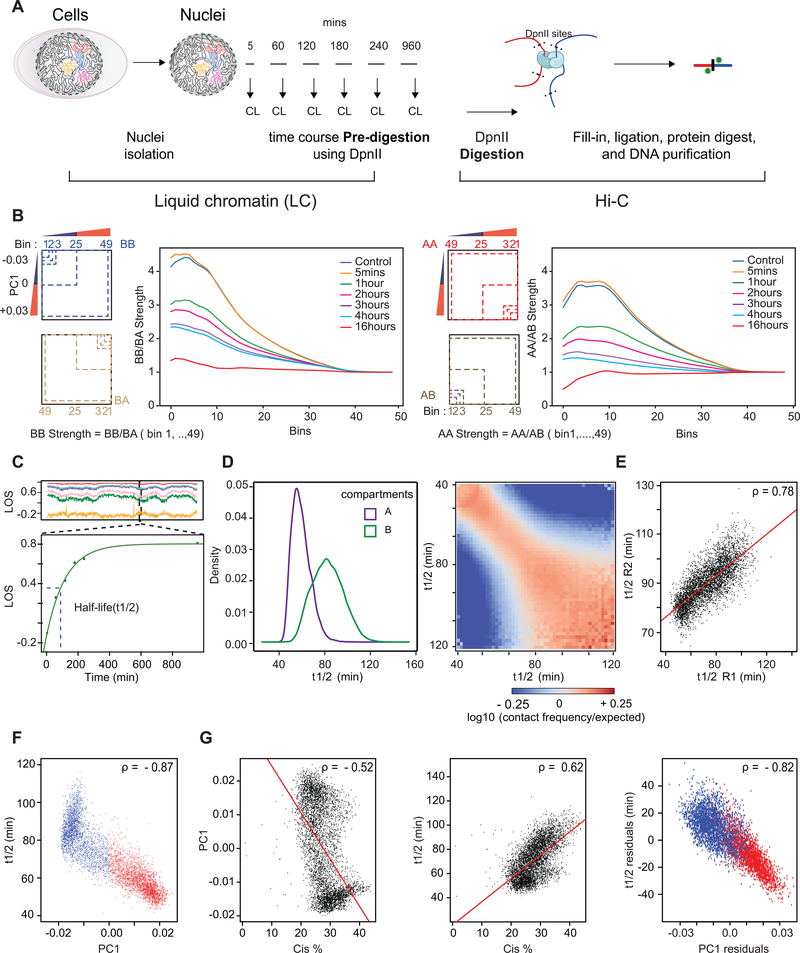
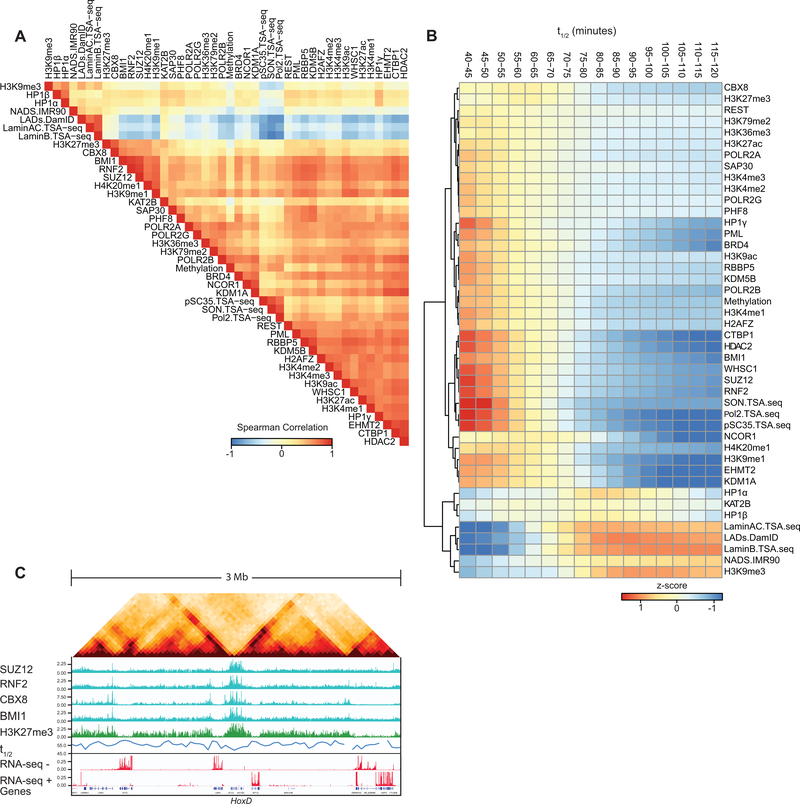
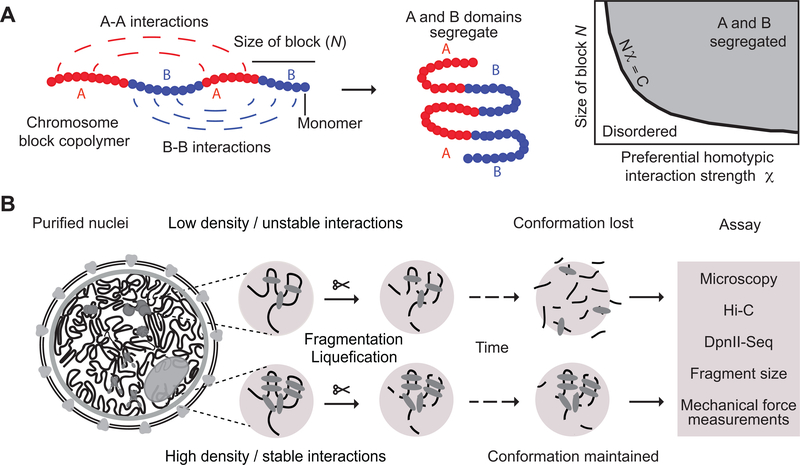
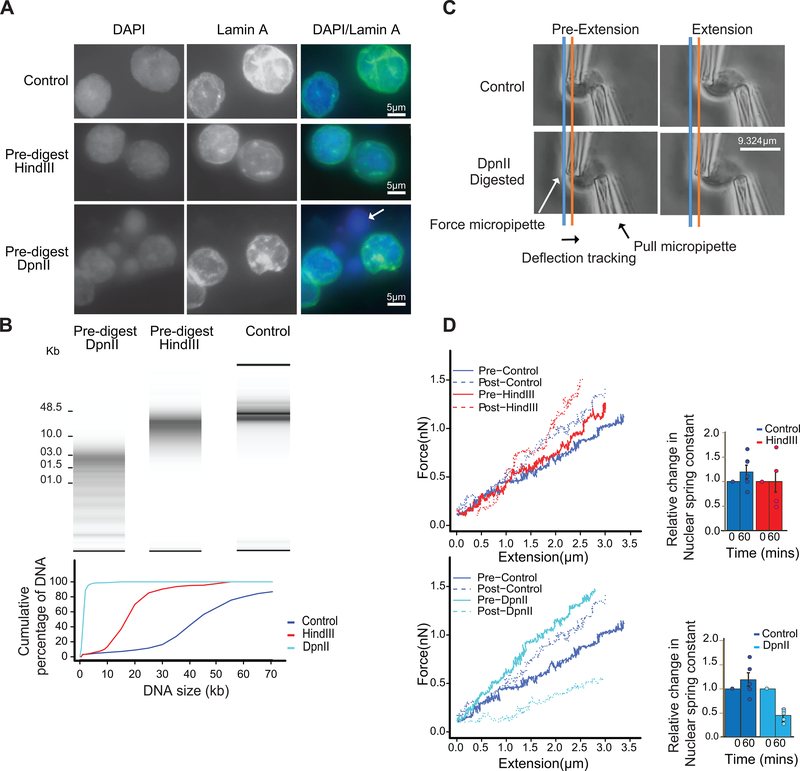
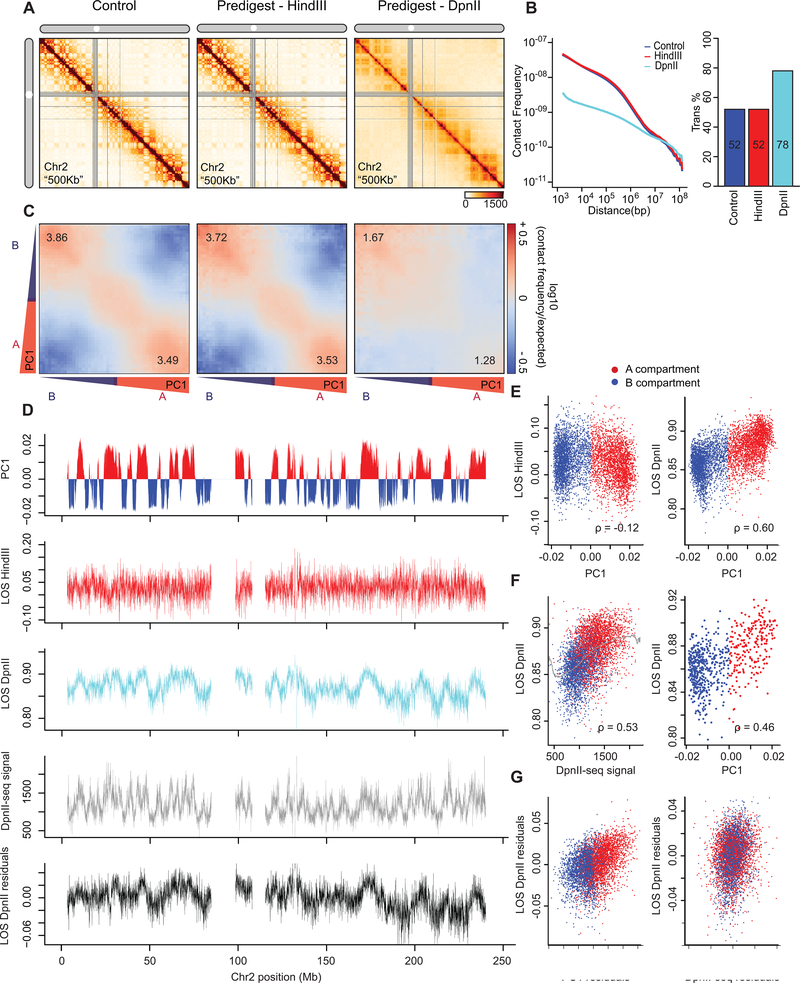
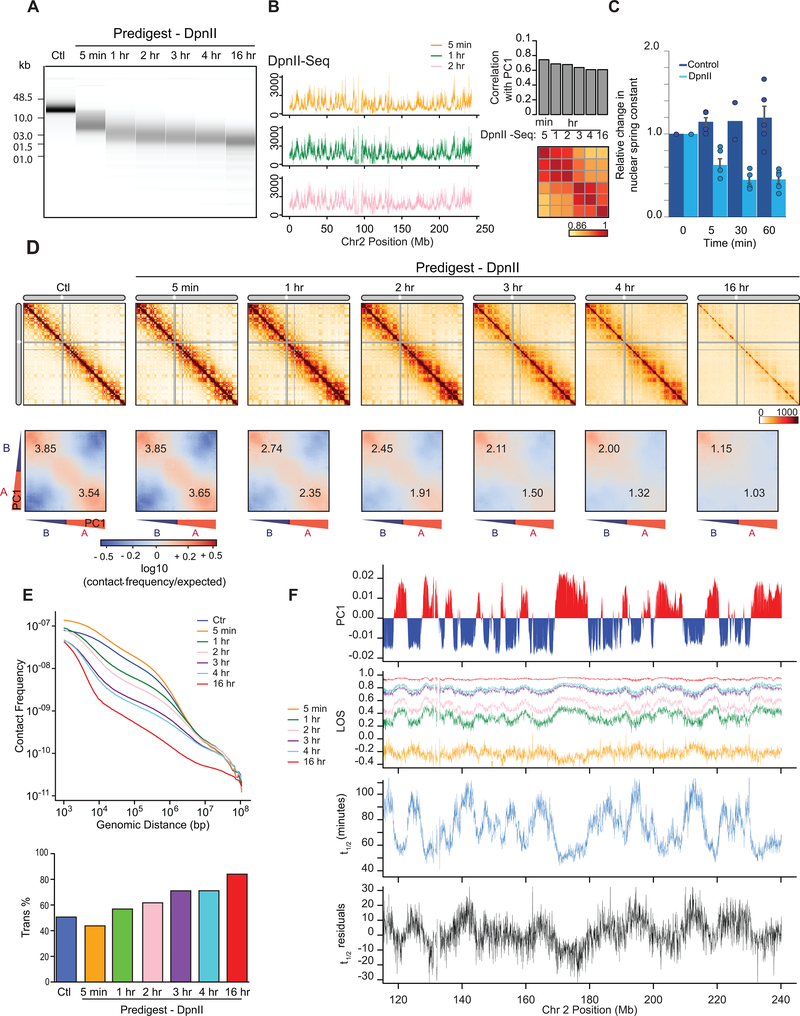
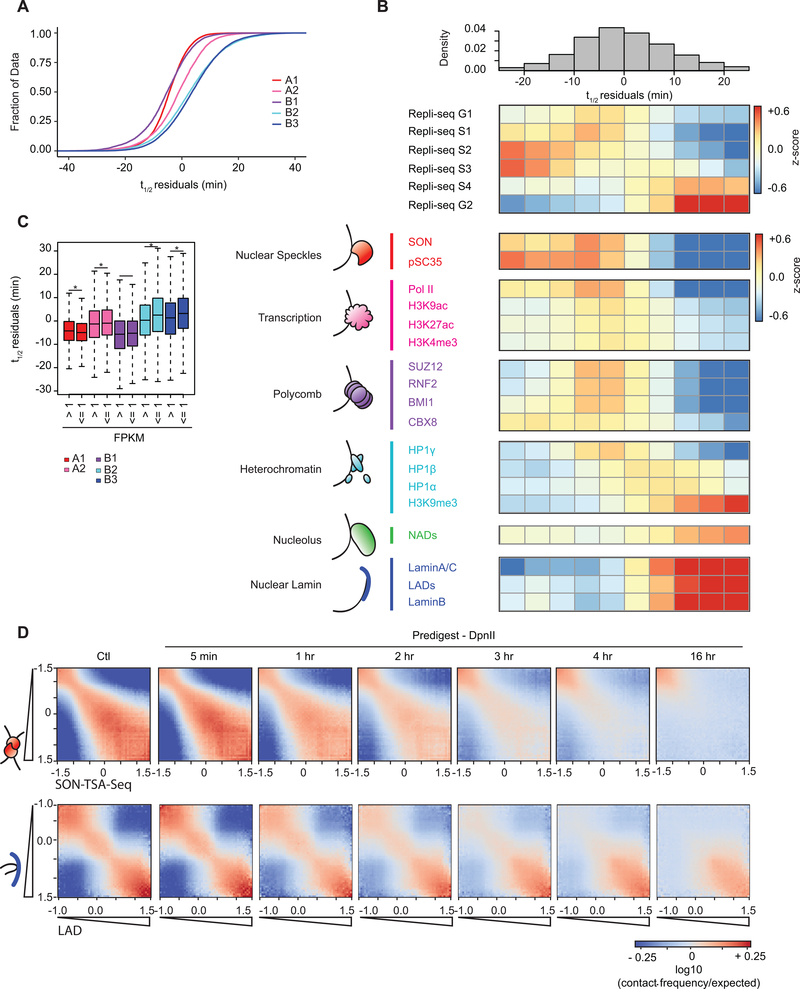
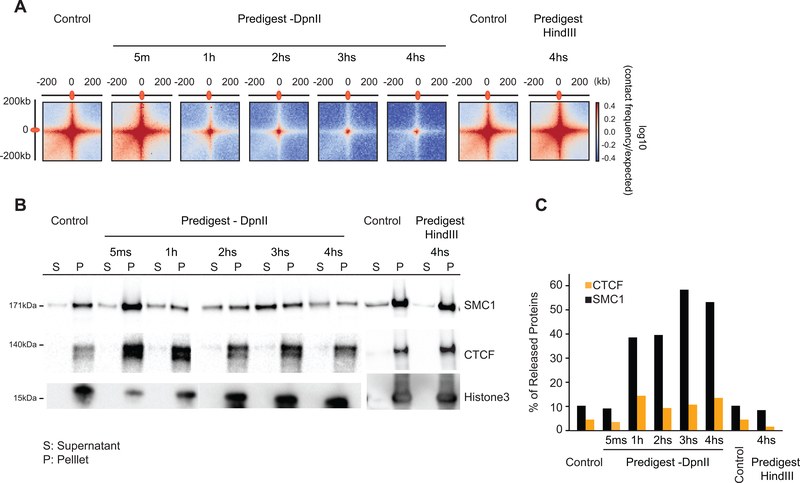
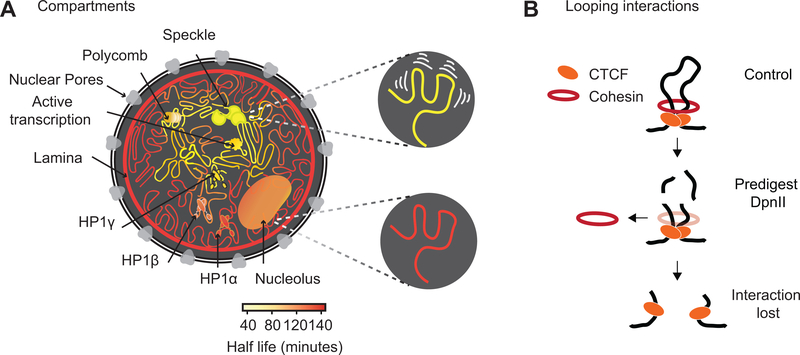
Nuclear compartmentalization of active and inactive chromatin is thought to occur through microphase separation mediated by interactions between loci of similar type. The nature and dynamics of these interactions are not known. We developed liquid chromatin Hi-C to map the stability of associations between loci. Before fixation and Hi-C, chromosomes are fragmented, which removes strong polymeric constraint, enabling detection of intrinsic locus-locus interaction stabilities. Compartmentalization is stable when fragments are larger than 10-25 kb. Fragmentation of chromatin into pieces smaller than 6 kb leads to gradual loss of genome organization. Lamin-associated domains are most stable, whereas interactions for speckle- and polycomb-associated loci are more dynamic. Cohesin-mediated loops dissolve after fragmentation. Liquid chromatin Hi-C provides a genome-wide view of chromosome interaction dynamics.
Conflict of interest statement
Competing interests statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
