

An interpretable low-complexity machine learning framework for robust exome-based in- silico diagnosis of Crohn's disease patients
- PMID: 33575557
- PMCID: PMC7671306
- DOI: 10.1093/nargab/lqaa011
An interpretable low-complexity machine learning framework for robust exome-based in- silico diagnosis of Crohn's disease patients
Abstract
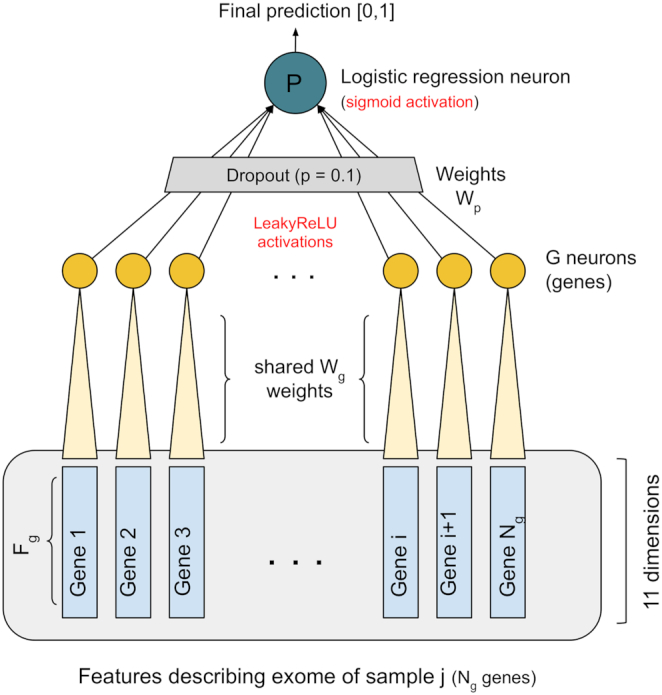
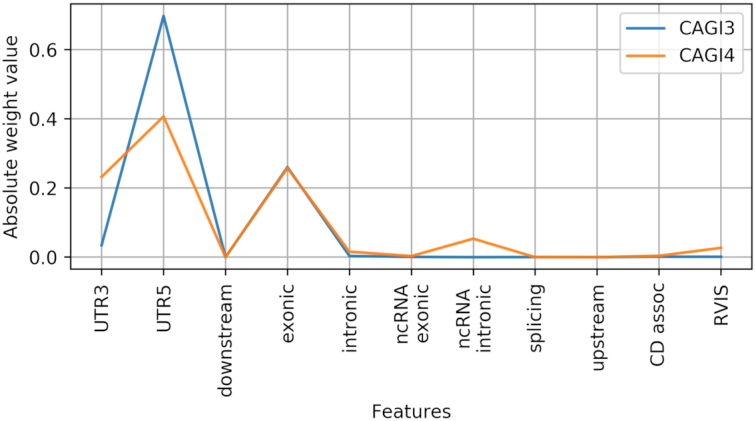
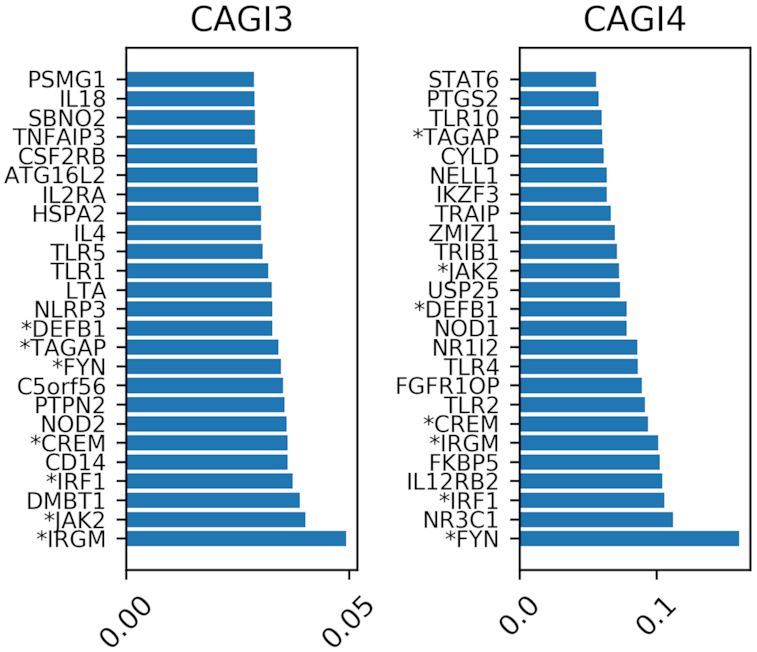
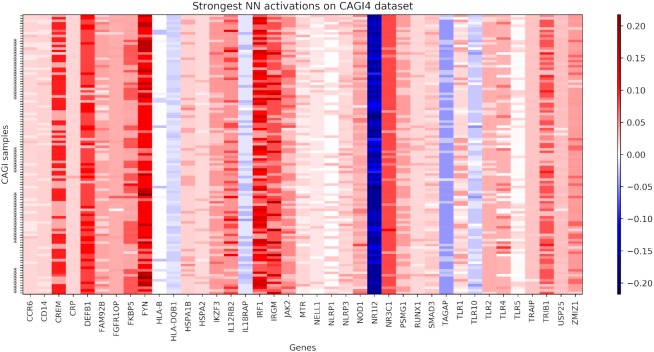
Whole exome sequencing (WES) data are allowing researchers to pinpoint the causes of many Mendelian disorders. In time, sequencing data will be crucial to solve the genome interpretation puzzle, which aims at uncovering the genotype-to-phenotype relationship, but for the moment many conceptual and technical problems need to be addressed. In particular, very few attempts at the in-silico diagnosis of oligo-to-polygenic disorders have been made so far, due to the complexity of the challenge, the relative scarcity of the data and issues such as batch effects and data heterogeneity, which are confounder factors for machine learning (ML) methods. Here, we propose a method for the exome-based in-silico diagnosis of Crohn's disease (CD) patients which addresses many of the current methodological issues. First, we devise a rational ML-friendly feature representation for WES data based on the gene mutational burden concept, which is suitable for small sample sizes datasets. Second, we propose a Neural Network (NN) with parameter tying and heavy regularization, in order to limit its complexity and thus the risk of over-fitting. We trained and tested our NN on 3 CD case-controls datasets, comparing the performance with the participants of previous CAGI challenges. We show that, notwithstanding the limited NN complexity, it outperforms the previous approaches. Moreover, we interpret the NN predictions by analyzing the learned patterns at the variant and gene level and investigating the decision process leading to each prediction.
© The Author(s) 2019. Published by Oxford University Press on behalf of NAR Genomics and Bioinformatics.
Figures
References
-
- Van Dijk E.L., Auger H., Jaszczyszyn Y., Thermes C.. Ten years of next-generation sequencing technology. Trends Genet. 2014; 30:418–426. - PubMed
-
- Bamshad M.J., Ng S.B., Bigham A.W., Tabor H.K., Emond M.J., Nickerson D.A., Shendure J.. Exome sequencing as a tool for Mendelian disease gene discovery. Nat. Rev. Genet. 2011; 12:745–755. - PubMed
-
- Boycott K.M., Vanstone M.R., Bulman D.E., MacKenzie A.E.. Rare-disease genetics in the era of next-generation sequencing: discovery to translation. Nat. Rev. Genet. 2013; 14:681–691. - PubMed
-
- Daneshjou R., Wang Y., Bromberg Y., Bovo S., Martelli P.L., Babbi G., Lena P.D., Casadio R., Edwards M., Gifford D. et al.. Working toward precision medicine: Predicting phenotypes from exomes in the Critical Assessment of Genome Interpretation (CAGI) challenges. Hum. Mutat. 2017; 38:1182–1192. - PMC - PubMed
LinkOut - more resources
Full Text Sources
