

Benchmarking of long-read correction methods
- PMID: 33575591
- PMCID: PMC7671305
- DOI: 10.1093/nargab/lqaa037
Benchmarking of long-read correction methods
Abstract
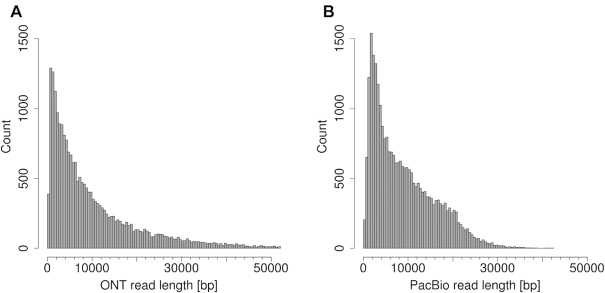
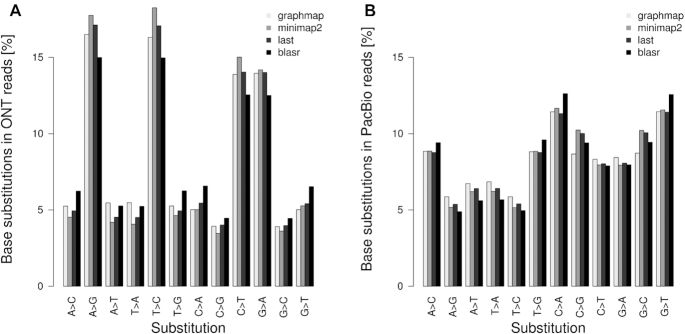
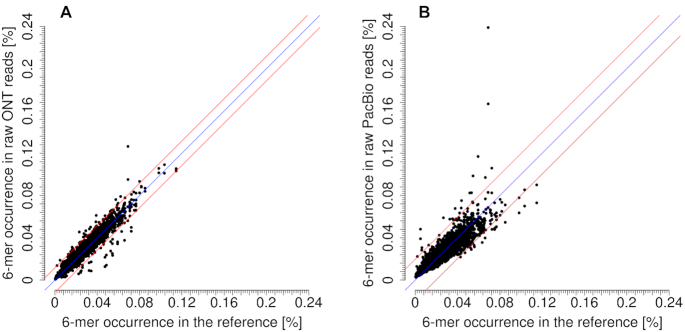
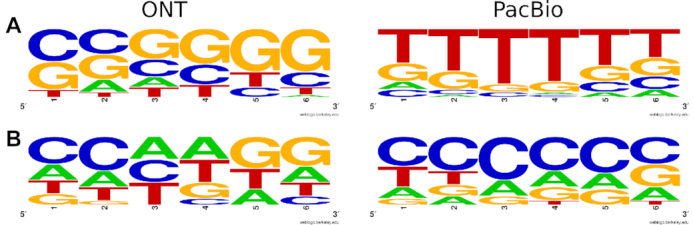
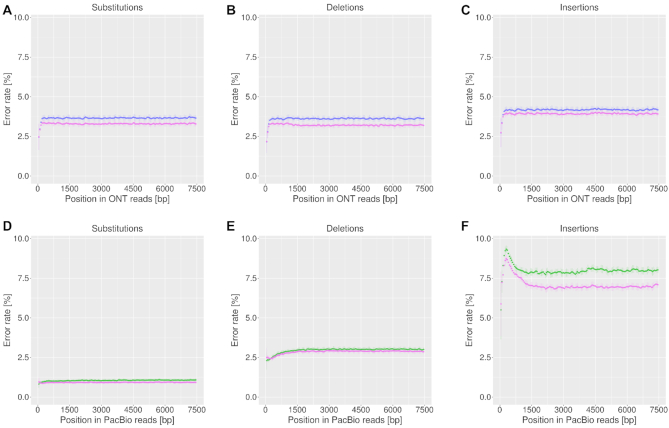
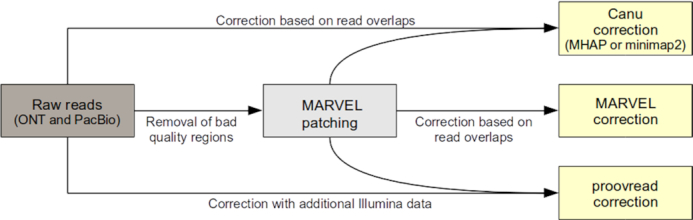
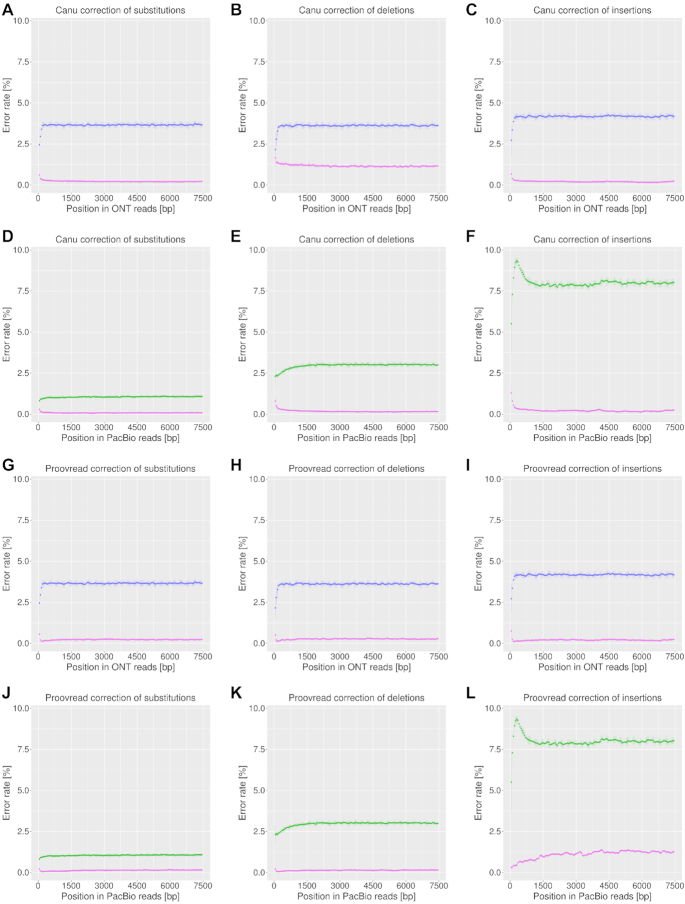
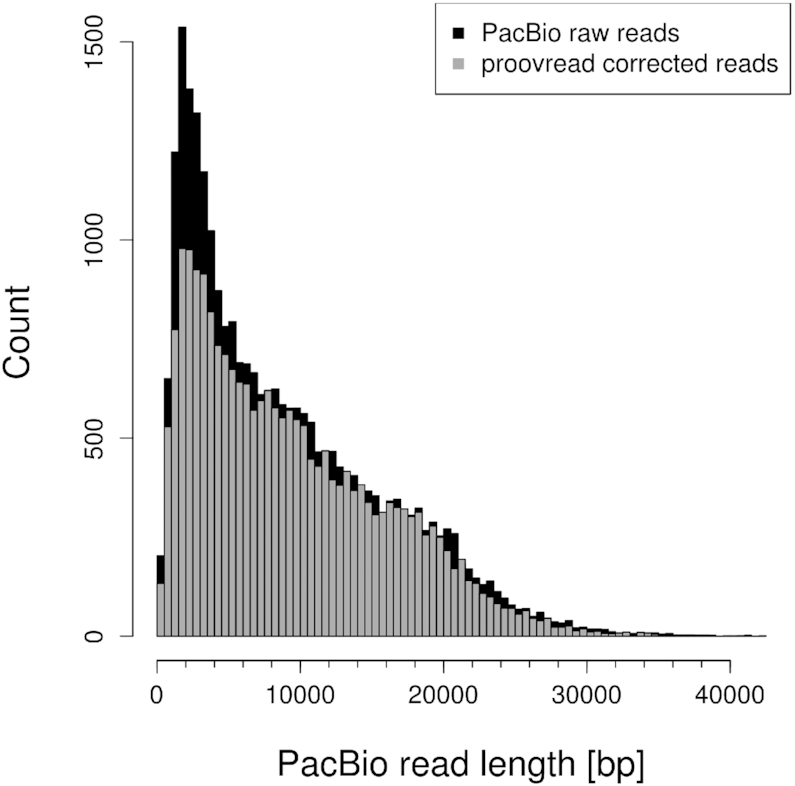
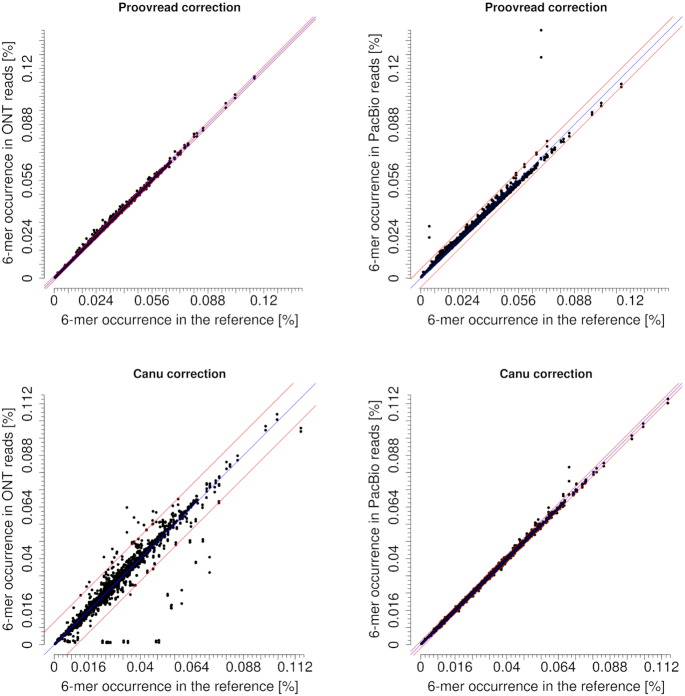
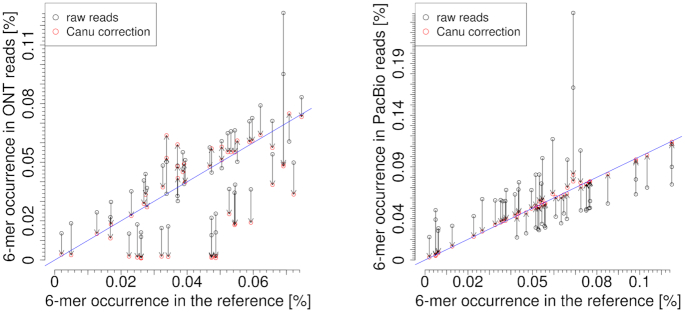
Third-generation sequencing technologies provided by Pacific Biosciences and Oxford Nanopore Technologies generate read lengths in the scale of kilobasepairs. However, these reads display high error rates, and correction steps are necessary to realize their great potential in genomics and transcriptomics. Here, we compare properties of PacBio and Nanopore data and assess correction methods by Canu, MARVEL and proovread in various combinations. We found total error rates of around 13% in the raw datasets. PacBio reads showed a high rate of insertions (around 8%) whereas Nanopore reads showed similar rates for substitutions, insertions and deletions of around 4% each. In data from both technologies the errors were uniformly distributed along reads apart from noisy 5' ends, and homopolymers appeared among the most over-represented kmers relative to a reference. Consensus correction using read overlaps reduced error rates to about 1% when using Canu or MARVEL after patching. The lowest error rate in Nanopore data (0.45%) was achieved by applying proovread on MARVEL-patched data including Illumina short-reads, and the lowest error rate in PacBio data (0.42%) was the result of Canu correction with minimap2 alignment after patching. Our study provides valuable insights and benchmarks regarding long-read data and correction methods.
© The Author(s) 2019. Published by Oxford University Press on behalf of NAR Genomics and Bioinformatics.
Figures

References
LinkOut - more resources
Full Text Sources
