

Fatal perinatal mitochondrial cardiac failure caused by recurrent de novo duplications in the ATAD3 locus
- PMID: 33575671
- PMCID: PMC7875323
- DOI: 10.1016/j.medj.2020.06.004
Fatal perinatal mitochondrial cardiac failure caused by recurrent de novo duplications in the ATAD3 locus
Abstract
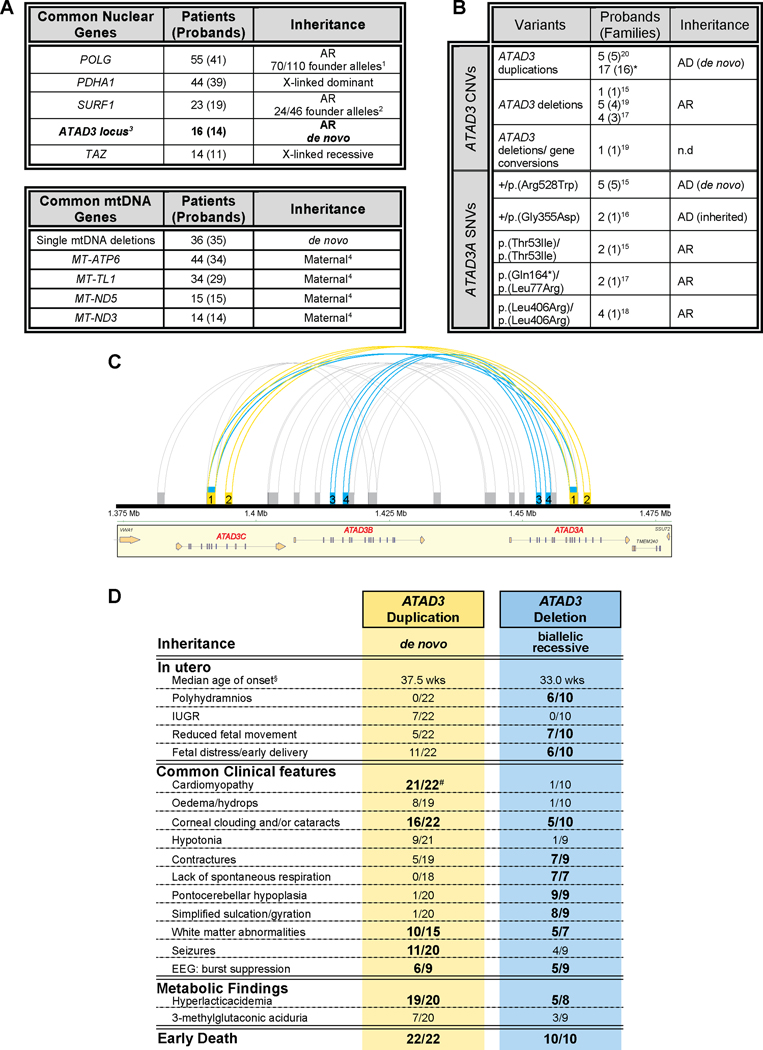
Background: In about half of all patients with a suspected monogenic disease, genomic investigations fail to identify the diagnosis. A contributing factor is the difficulty with repetitive regions of the genome, such as those generated by segmental duplications. The ATAD3 locus is one such region, in which recessive deletions and dominant duplications have recently been reported to cause lethal perinatal mitochondrial diseases characterized by pontocerebellar hypoplasia or cardiomyopathy, respectively.
Methods: Whole exome, whole genome and long-read DNA sequencing techniques combined with studies of RNA and quantitative proteomics were used to investigate 17 subjects from 16 unrelated families with suspected mitochondrial disease.
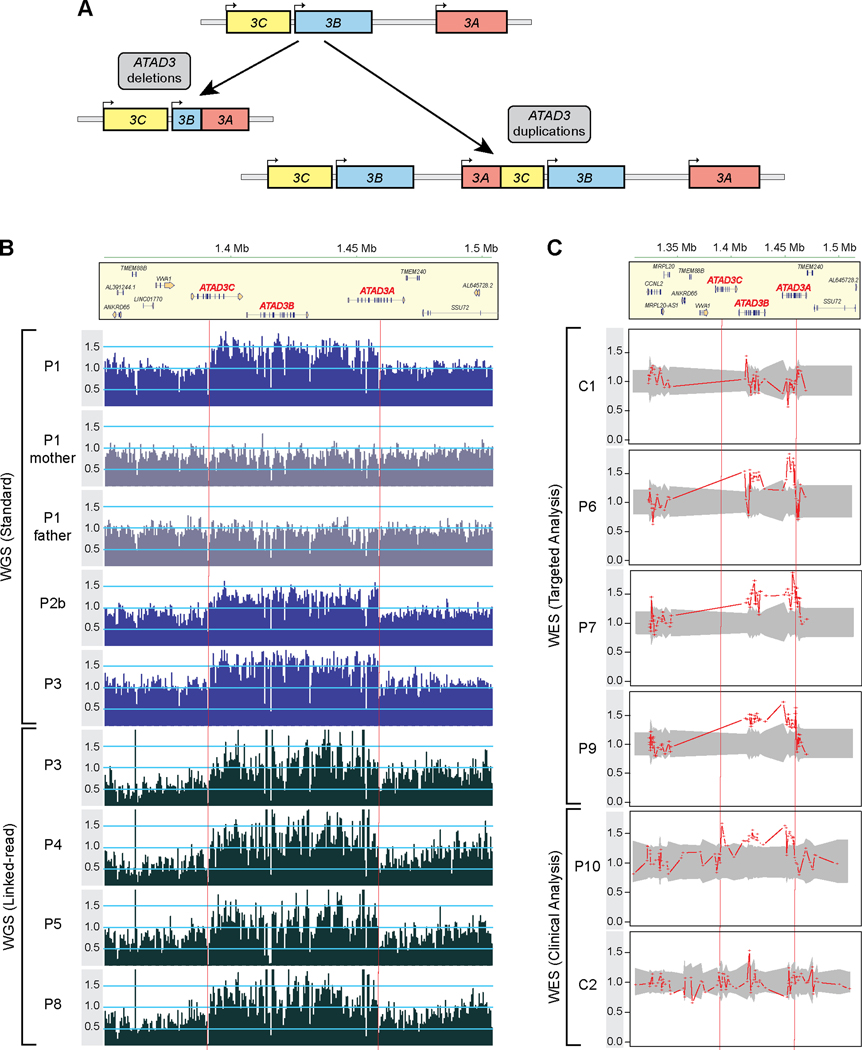
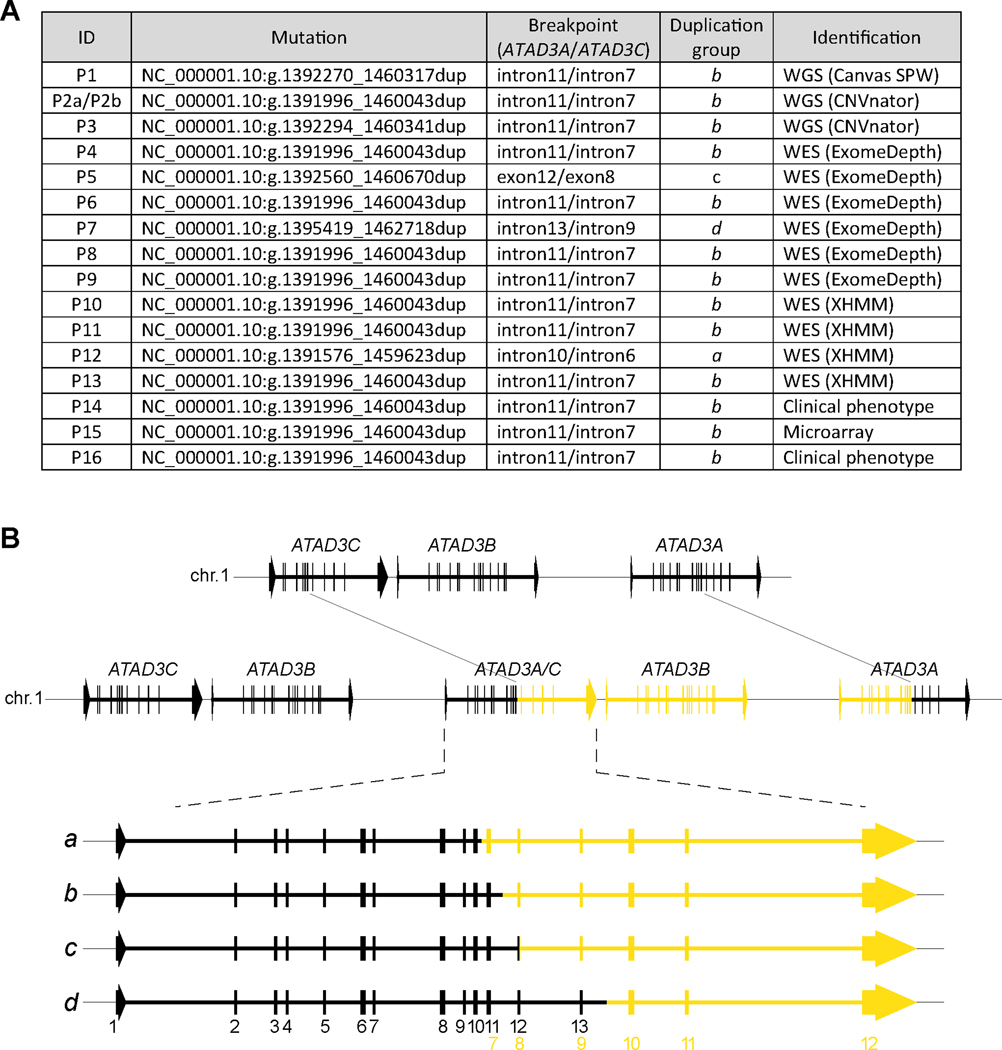
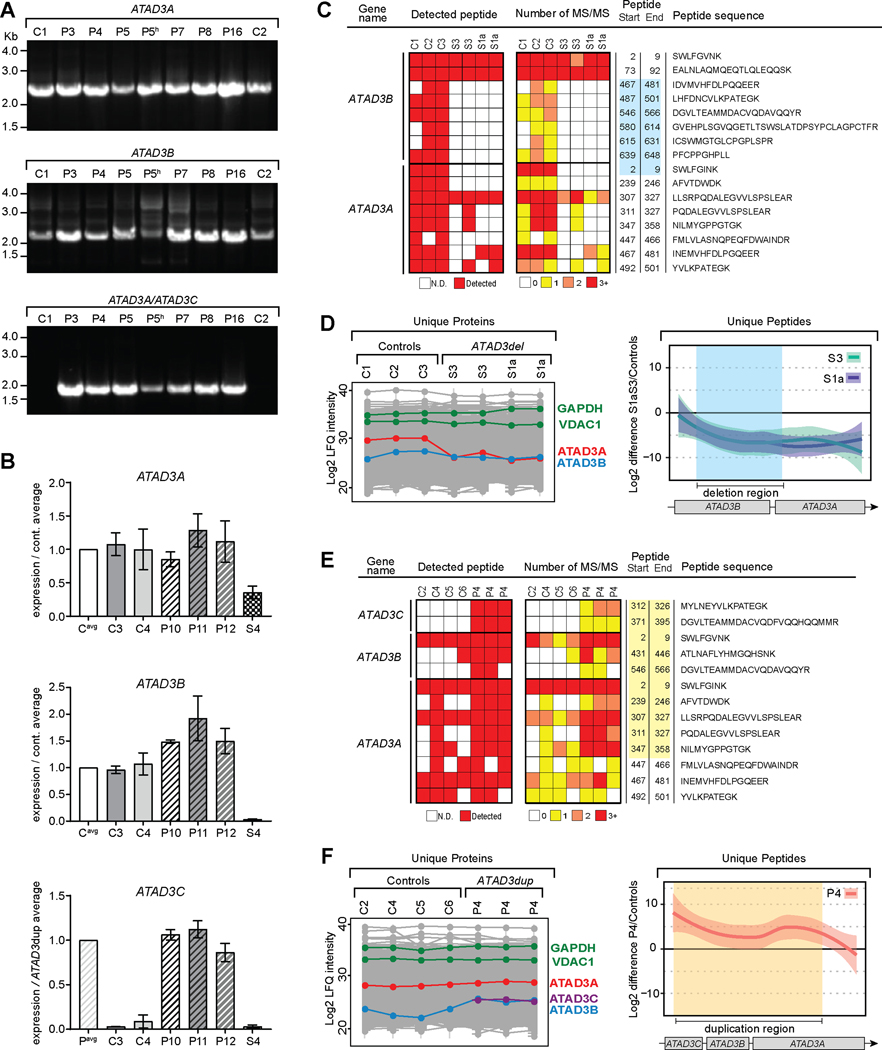
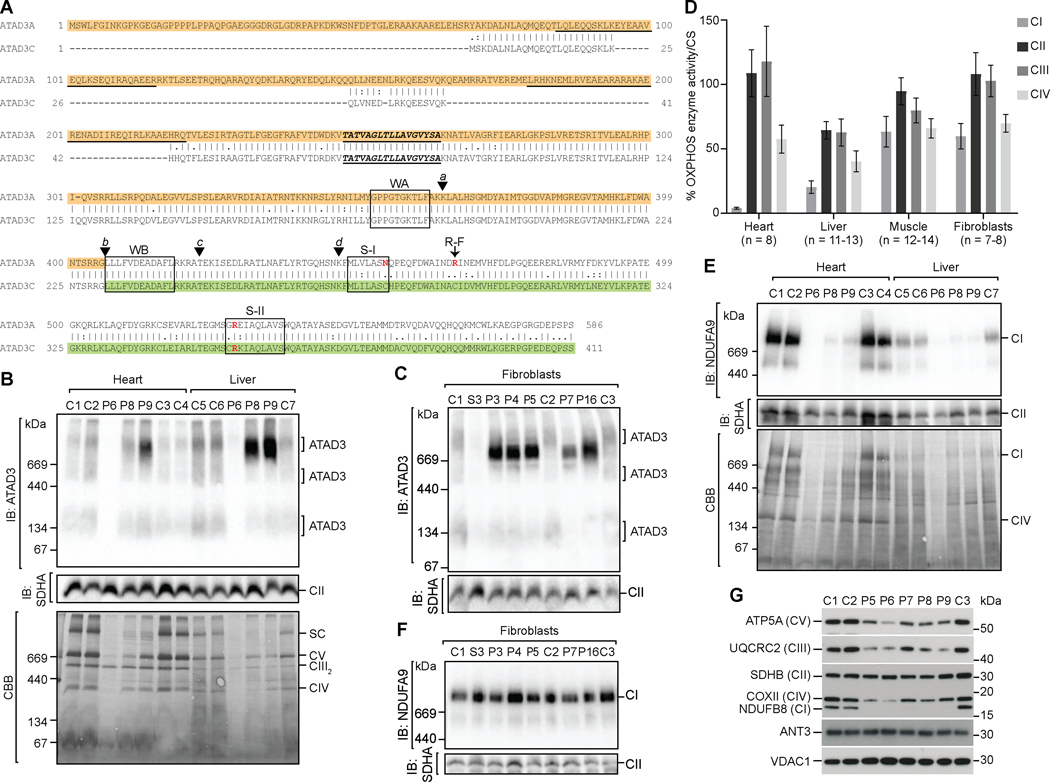
Findings: We report six different de novo duplications in the ATAD3 gene locus causing a distinctive presentation including lethal perinatal cardiomyopathy, persistent hyperlactacidemia, and frequently corneal clouding or cataracts and encephalopathy. The recurrent 68 Kb ATAD3 duplications are identifiable from genome and exome sequencing but usually missed by microarrays. The ATAD3 duplications result in the formation of identical chimeric ATAD3A/ATAD3C proteins, altered ATAD3 complexes and a striking reduction in mitochondrial oxidative phosphorylation complex I and its activity in heart tissue.
Conclusions: ATAD3 duplications appear to act in a dominant-negative manner and the de novo inheritance infers a low recurrence risk for families, unlike most pediatric mitochondrial diseases. More than 350 genes underlie mitochondrial diseases. In our experience the ATAD3 locus is now one of the five most common causes of nuclear-encoded pediatric mitochondrial disease but the repetitive nature of the locus means ATAD3 diagnoses may be frequently missed by current genomic strategies.
Funding: Australian NHMRC, US Department of Defense, Japanese AMED and JSPS agencies, Australian Genomics Health Alliance and Australian Mito Foundation.
Keywords: ATAD3; cardiomyopathy; genomics; mitochondrial disease; quantitative proteomics; segmental duplication.
Figures
Comment in
-
Cutting the Gordian Knot of a Mitochondrial Disease.Med. 2021 Jan 15;2(1):11-13. doi: 10.1016/j.medj.2020.12.015. Med. 2021. PMID: 35590127
References
-
- Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, Suomalainen A, Thorburn DR, Zeviani M, and Turnbull DM (2016). Mitochondrial diseases. Nat Rev Dis Primers 2, 16080. - PubMed
-
- Rahman J, and Rahman S. (2018). Mitochondrial medicine in the omics era. Lancet 391, 2560–2574. - PubMed
-
- Stenton SL, and Prokisch H. (2018). Advancing genomic approaches to the molecular diagnosis of mitochondrial disease. Essays Biochem 62, 399–408. - PubMed
-
- Stankiewicz P, and Lupski JR (2002). Genome architecture, rearrangements and genomic disorders. Trends Genet 18, 74–82. - PubMed
-
- Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, Schwartz S, Adams MD, Myers EW, Li PW, and Eichler EE (2002). Recent segmental duplications in the human genome. Science 297, 1003–1007. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical