

Proteogenomic and metabolomic characterization of human glioblastoma
- PMID: 33577785
- PMCID: PMC8044053
- DOI: 10.1016/j.ccell.2021.01.006
Proteogenomic and metabolomic characterization of human glioblastoma
Abstract
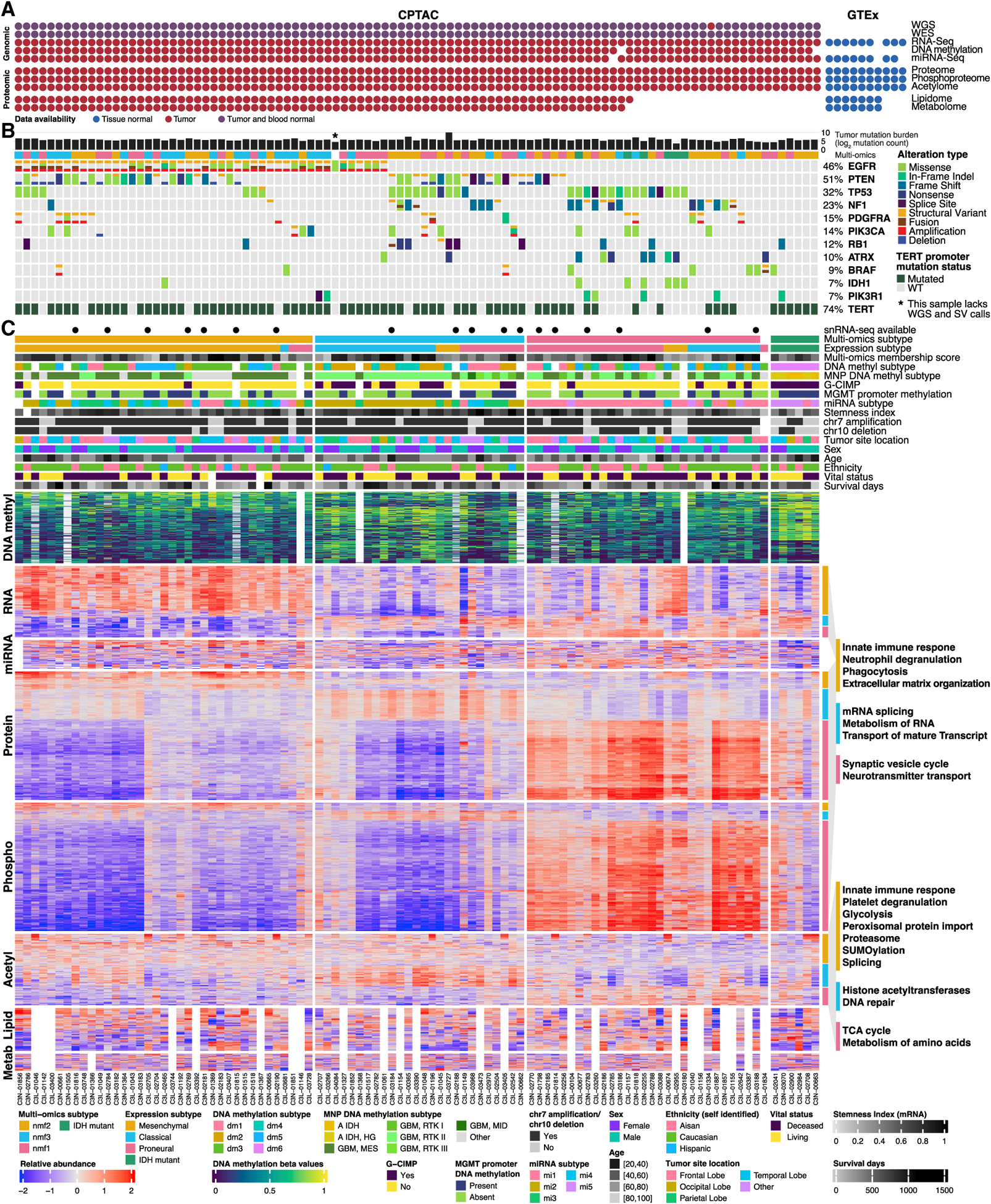
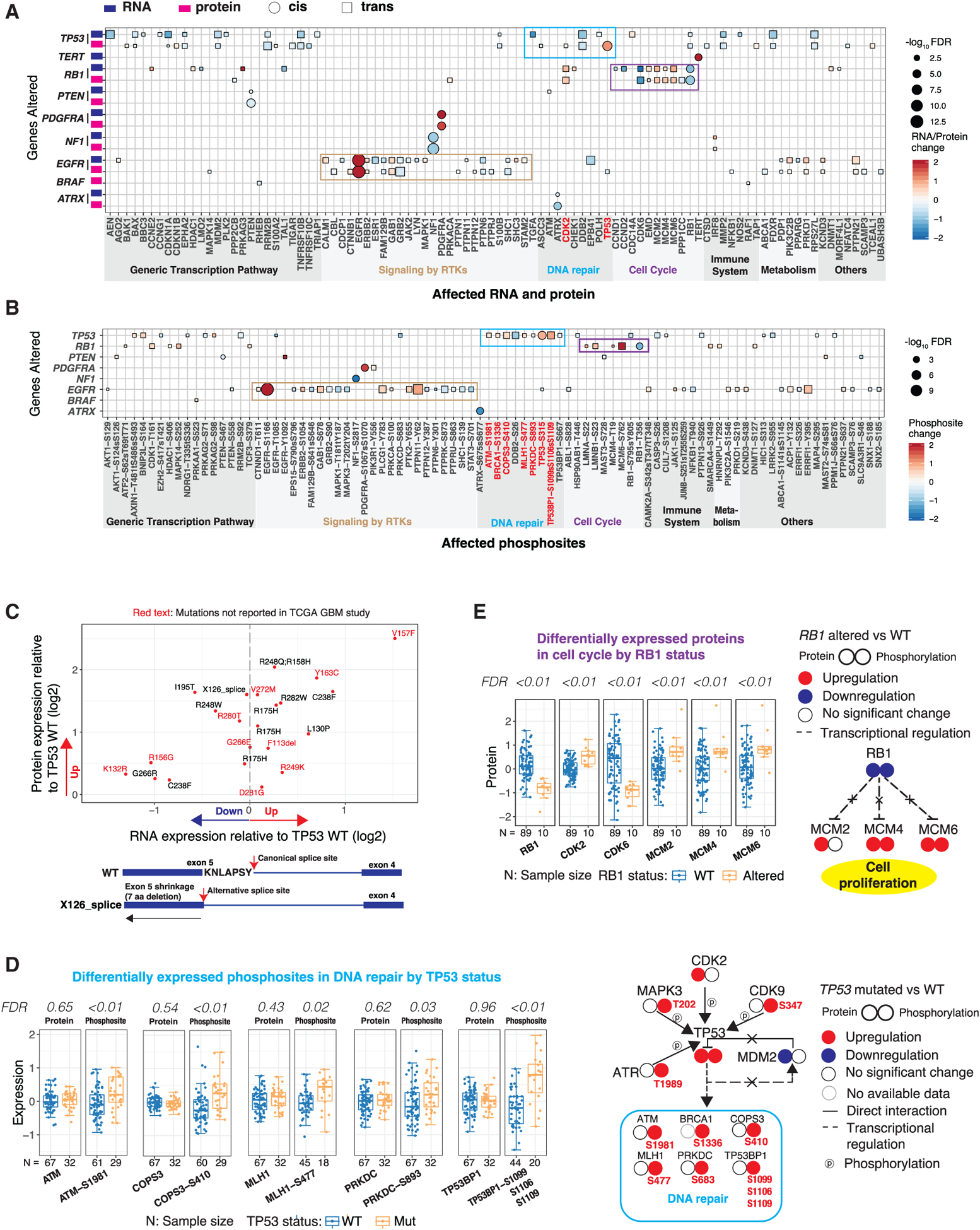
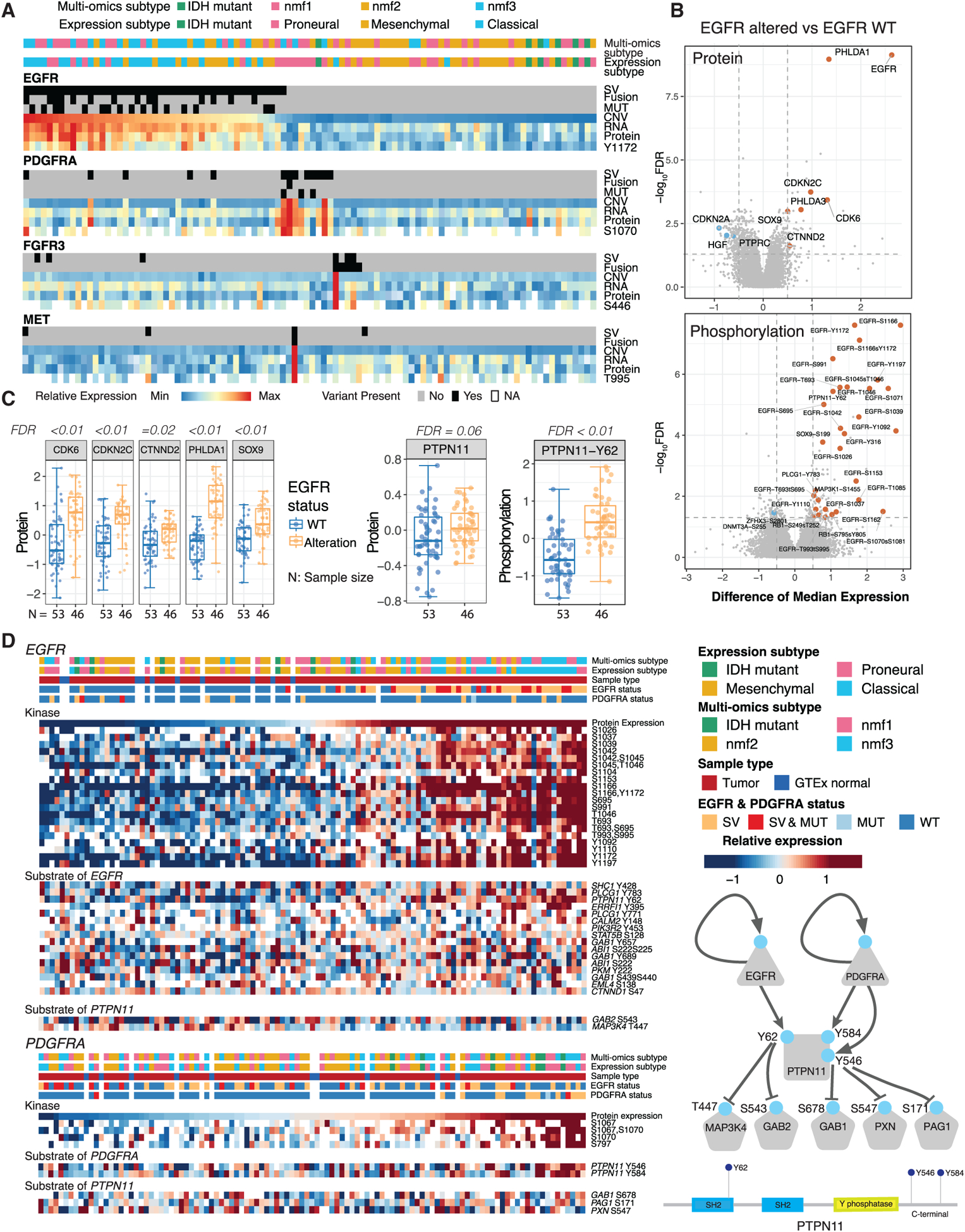
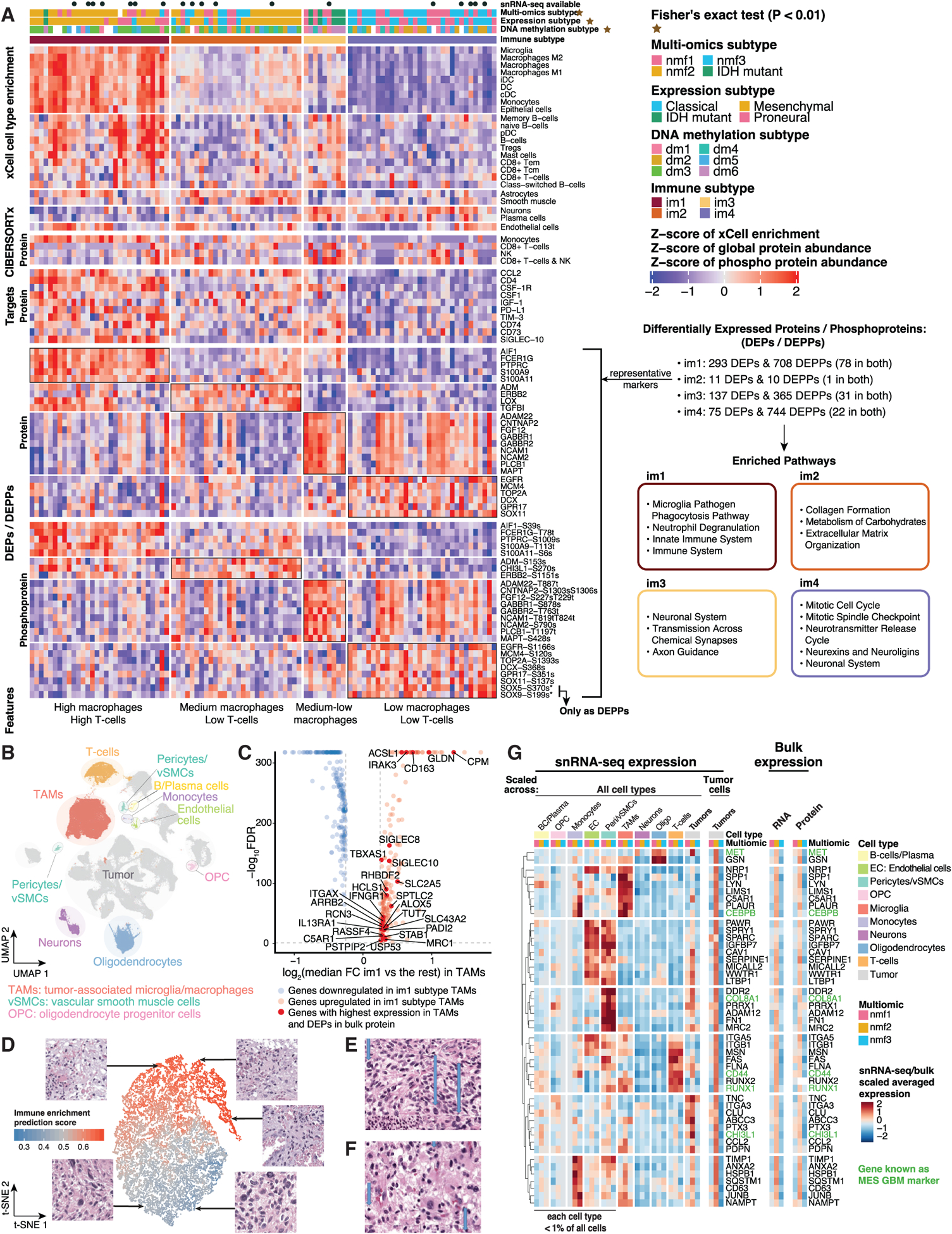
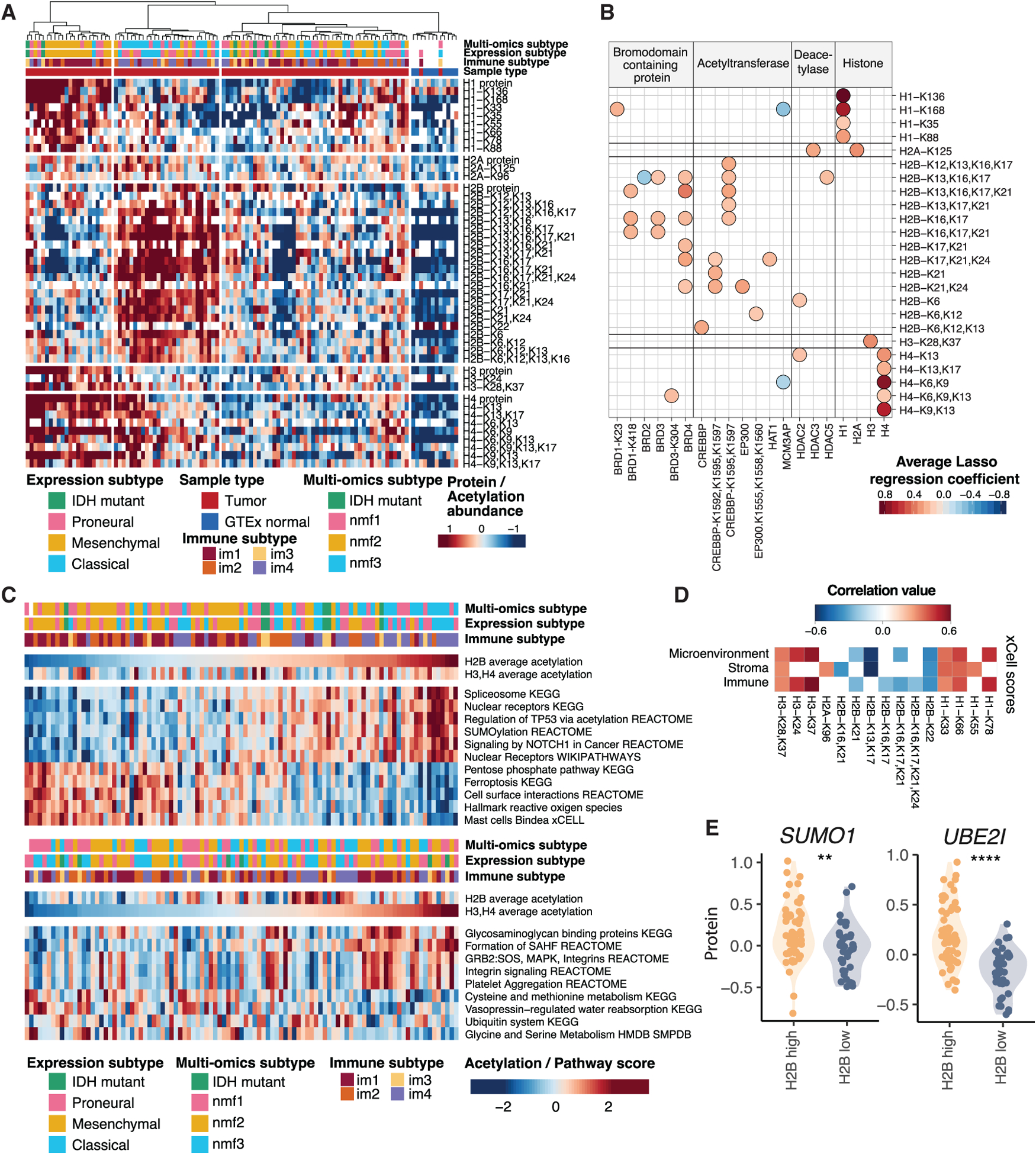
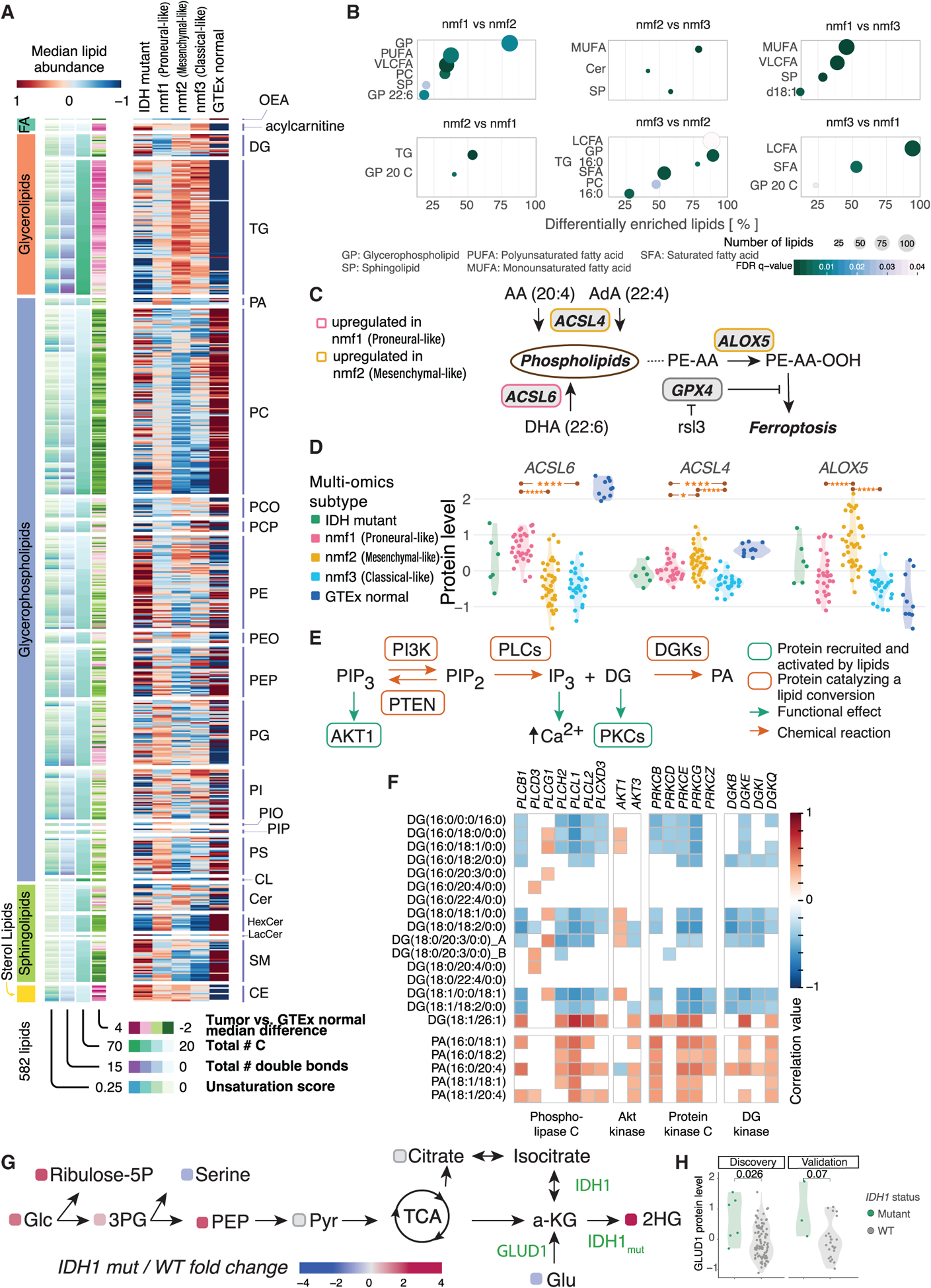
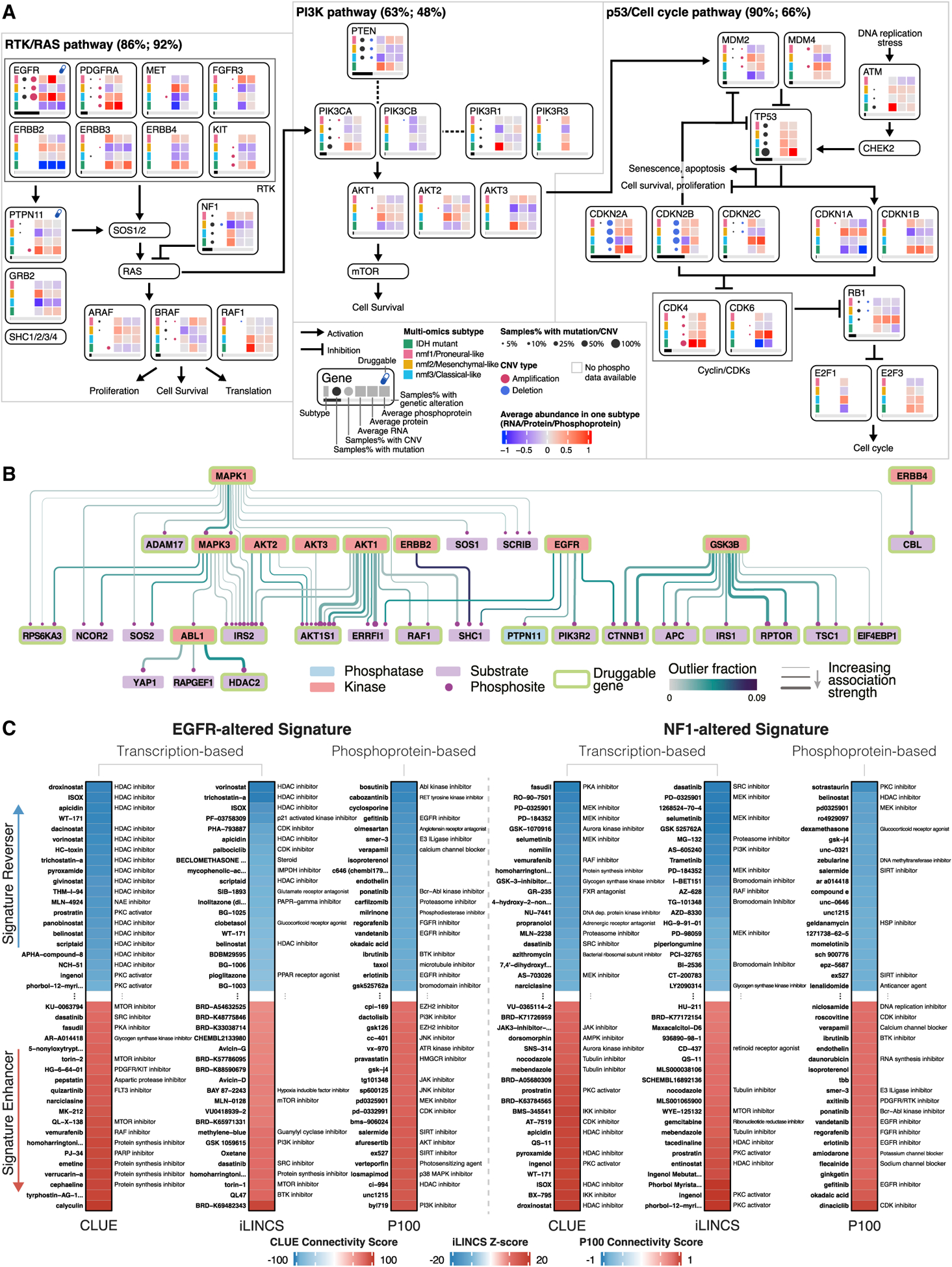
Glioblastoma (GBM) is the most aggressive nervous system cancer. Understanding its molecular pathogenesis is crucial to improving diagnosis and treatment. Integrated analysis of genomic, proteomic, post-translational modification and metabolomic data on 99 treatment-naive GBMs provides insights to GBM biology. We identify key phosphorylation events (e.g., phosphorylated PTPN11 and PLCG1) as potential switches mediating oncogenic pathway activation, as well as potential targets for EGFR-, TP53-, and RB1-altered tumors. Immune subtypes with distinct immune cell types are discovered using bulk omics methodologies, validated by snRNA-seq, and correlated with specific expression and histone acetylation patterns. Histone H2B acetylation in classical-like and immune-low GBM is driven largely by BRDs, CREBBP, and EP300. Integrated metabolomic and proteomic data identify specific lipid distributions across subtypes and distinct global metabolic changes in IDH-mutated tumors. This work highlights biological relationships that could contribute to stratification of GBM patients for more effective treatment.
Keywords: CPTAC; acetylome; glioblastoma; lipidome; metabolome; proteogenomics; proteomics; signaling; single nuclei RNA-seq.
Copyright © 2021 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests S.Y. is employed by Sema4. A.H.K. consults for Monteris Medical. P.W. is a statistical consultant for Sema4. M.G.C. receives research support from Orbus Therapeutics and NeoimmuneTech Inc, and royalties from UpToDate.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- U24 CA210954/CA/NCI NIH HHS/United States
- P30 ES010126/ES/NIEHS NIH HHS/United States
- U24 CA210985/CA/NCI NIH HHS/United States
- U24 CA210967/CA/NCI NIH HHS/United States
- U24 CA210986/CA/NCI NIH HHS/United States
- T32 HG000045/HG/NHGRI NIH HHS/United States
- U24 CA210972/CA/NCI NIH HHS/United States
- U24 CA210955/CA/NCI NIH HHS/United States
- K08 NS102474/NS/NINDS NIH HHS/United States
- U24 CA210979/CA/NCI NIH HHS/United States
- U01 CA214125/CA/NCI NIH HHS/United States
- R01 HG009711/HG/NHGRI NIH HHS/United States
- U24 CA160019/CA/NCI NIH HHS/United States
- R01 NS107833/NS/NINDS NIH HHS/United States
- U24 CA210993/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
