

Prostaglandin E2 promotes intestinal inflammation via inhibiting microbiota-dependent regulatory T cells
- PMID: 33579710
- PMCID: PMC7880593
- DOI: 10.1126/sciadv.abd7954
Prostaglandin E2 promotes intestinal inflammation via inhibiting microbiota-dependent regulatory T cells
Abstract
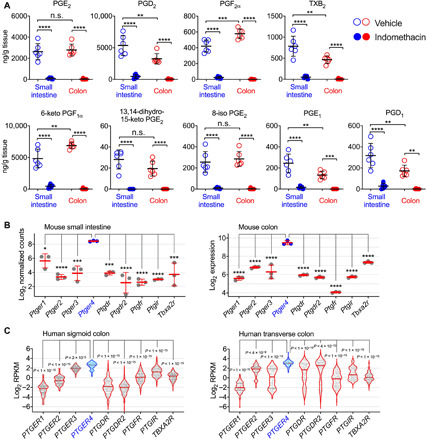
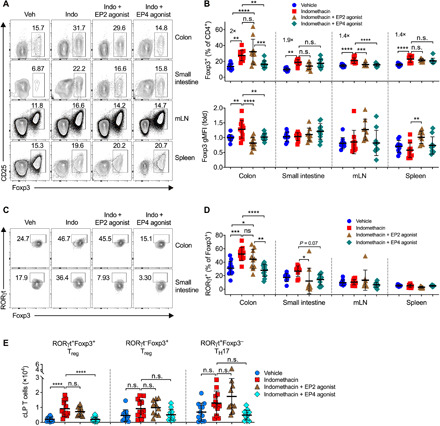
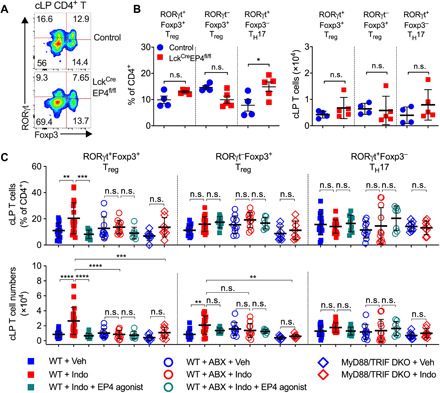
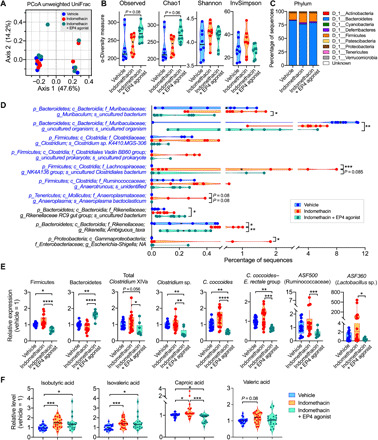
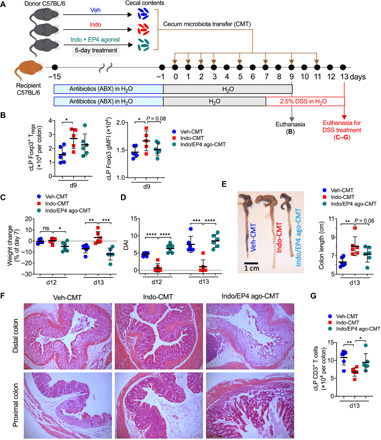
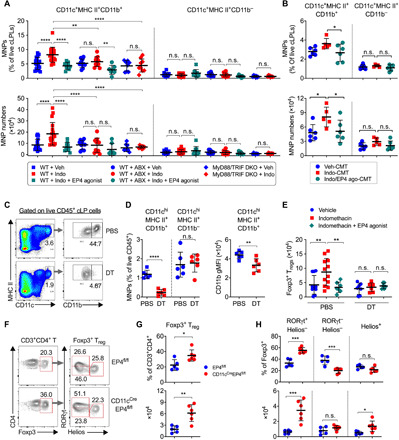
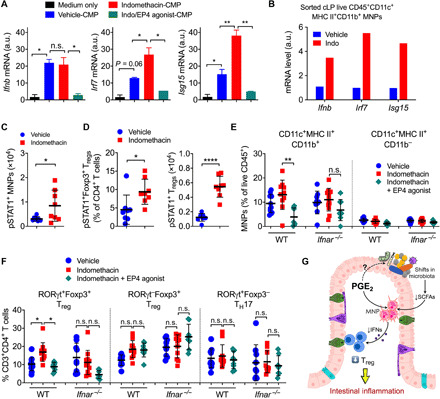
The gut microbiota fundamentally regulates intestinal homeostasis and disease partially through mechanisms that involve modulation of regulatory T cells (Tregs), yet how the microbiota-Treg cross-talk is physiologically controlled is incompletely defined. Here, we report that prostaglandin E2 (PGE2), a well-known mediator of inflammation, inhibits mucosal Tregs in a manner depending on the gut microbiota. PGE2 through its receptor EP4 diminishes Treg-favorable commensal microbiota. Transfer of the gut microbiota that was modified by PGE2-EP4 signaling modulates mucosal Treg responses and exacerbates intestinal inflammation. Mechanistically, PGE2-modified microbiota regulates intestinal mononuclear phagocytes and type I interferon signaling. Depletion of mononuclear phagocytes or deficiency of type I interferon receptor diminishes PGE2-dependent Treg inhibition. Together, our findings provide emergent evidence that PGE2-mediated disruption of microbiota-Treg communication fosters intestinal inflammation.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works. Distributed under a Creative Commons Attribution License 4.0 (CC BY).
Figures
Similar articles
-
Prostaglandin E2 directly inhibits the conversion of inducible regulatory T cells through EP2 and EP4 receptors via antagonizing TGF-β signalling.Immunology. 2021 Dec;164(4):777-791. doi: 10.1111/imm.13417. Epub 2021 Oct 1. Immunology. 2021. PMID: 34529833 Free PMC article.
-
Prostaglandin E2 restrains human Treg cell differentiation via E prostanoid receptor 2-protein kinase A signaling.Immunol Lett. 2017 Nov;191:63-72. doi: 10.1016/j.imlet.2017.09.009. Epub 2017 Sep 28. Immunol Lett. 2017. PMID: 28963072
-
Metabolic regulation by prostaglandin E2 impairs lung group 2 innate lymphoid cell responses.Allergy. 2023 Mar;78(3):714-730. doi: 10.1111/all.15541. Epub 2022 Oct 14. Allergy. 2023. PMID: 36181709 Free PMC article.
-
Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling.J Exp Med. 2009 Mar 16;206(3):535-48. doi: 10.1084/jem.20082293. Epub 2009 Mar 9. J Exp Med. 2009. PMID: 19273625 Free PMC article.
-
Prostaglandin E2-induced inflammation: Relevance of prostaglandin E receptors.Biochim Biophys Acta. 2015 Apr;1851(4):414-21. doi: 10.1016/j.bbalip.2014.07.008. Epub 2014 Jul 17. Biochim Biophys Acta. 2015. PMID: 25038274 Review.
Cited by
-
Electrolyzed Hydrogen Water Alleviates Abdominal Pain through Suppression of Colonic Tissue Inflammation in a Rat Model of Inflammatory Bowel Disease.Nutrients. 2022 Oct 22;14(21):4451. doi: 10.3390/nu14214451. Nutrients. 2022. PMID: 36364715 Free PMC article.
-
Low-Dose Aspirin Use in Pregnancy in Patients with IBD with Risk Factors for Preeclampsia: A Retrospective Cohort Study.Dig Dis Sci. 2024 Aug;69(8):2719-2720. doi: 10.1007/s10620-024-08544-0. Epub 2024 Jul 4. Dig Dis Sci. 2024. PMID: 38965156 No abstract available.
-
Gut Microbiota: An Important Player in Type 2 Diabetes Mellitus.Front Cell Infect Microbiol. 2022 Feb 15;12:834485. doi: 10.3389/fcimb.2022.834485. eCollection 2022. Front Cell Infect Microbiol. 2022. PMID: 35242721 Free PMC article. Review.
-
A Review of Gut Microbiota-Derived Metabolites in Tumor Progression and Cancer Therapy.Adv Sci (Weinh). 2023 May;10(15):e2207366. doi: 10.1002/advs.202207366. Epub 2023 Mar 23. Adv Sci (Weinh). 2023. PMID: 36951547 Free PMC article. Review.
-
L-Theanine Mitigates Chronic Alcoholic Intestinal Injury by Regulating Intestinal Alcohol and Linoleic-Arachidonic Acid Metabolism in Rats.Nutrients. 2025 Jun 5;17(11):1943. doi: 10.3390/nu17111943. Nutrients. 2025. PMID: 40507212 Free PMC article.
References
-
- Uhlig H. H., Powrie F., Translating immunology into therapeutic concepts for inflammatory bowel disease. Annu. Rev. Immunol. 36, 755–781 (2018). - PubMed
-
- Lynch S. V., Pedersen O., The human intestinal microbiome in health and disease. N. Engl. J. Med. 375, 2369–2379 (2016). - PubMed
-
- Honda K., Littman D. R., The microbiota in adaptive immune homeostasis and disease. Nature 535, 75–84 (2016). - PubMed
-
- Atarashi K., Tanoue T., Oshima K., Suda W., Nagano Y., Nishikawa H., Fukuda S., Saito T., Narushima S., Hase K., Kim S., Fritz J. V., Wilmes P., Ueha S., Matsushima K., Ohno H., Olle B., Sakaguchi S., Taniguchi T., Morita H., Hattori M., Honda K., Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 500, 232–236 (2013). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
