

Huntington's Disease Pathogenesis: Two Sequential Components
- PMID: 33579862
- PMCID: PMC7990433
- DOI: 10.3233/JHD-200427
Huntington's Disease Pathogenesis: Two Sequential Components
Abstract
Historically, Huntington's disease (HD; OMIM #143100) has played an important role in the enormous advances in human genetics seen over the past four decades. This familial neurodegenerative disorder involves variable onset followed by consistent worsening of characteristic abnormal movements along with cognitive decline and psychiatric disturbances. HD was the first autosomal disease for which the genetic defect was assigned to a position on the human chromosomes using only genetic linkage analysis with common DNA polymorphisms. This discovery set off a multitude of similar studies in other diseases, while the HD gene, later renamed HTT, and its vicinity in chromosome 4p16.3 then acted as a proving ground for development of technologies to clone and sequence genes based upon their genomic location, with the growing momentum of such advances fueling the Human Genome Project. The identification of the HD gene has not yet led to an effective treatment, but continued human genetic analysis of genotype-phenotype relationships in large HD subject populations, first at the HTT locus and subsequently genome-wide, has provided insights into pathogenesis that divide the course of the disease into two sequential, mechanistically distinct components.
Keywords: Huntington disease; genetic association; genetics; genotype-phenotype correlation; modifier gene; trinucleotide repeat expansion.
Conflict of interest statement
E.P.H., M.E.M, P.H., M.O., S.K. and J.M.L. have no disclosures.
V.C.W. is a scientific advisory board member of Triplet Therapeutics, a company developing new therapeutic approaches to address triplet repeat disorders such Huntington’s disease and Myotonic Dystrophy. Her financial interests in Triplet Therapeutics were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. She is a scientific advisory board member of LoQus23 Therapeutics and has provided paid consulting services to Alnylam.
L.J. is a member of the scientific advisory boards of Triplet Therapeutics and LoQus23 Therapeutics.
D.G.M. has been a scientific consultant and/or received honoraria or stock options from Biogen Idec, AMO Pharma, Charles River, Vertex Pharmaceuticals, Triplet Therapeutics, LoQus23, and Small Molecule RNA and has had research contracts with AMO Pharma and Vertex Pharmaceuticals.
J.D.L. is a paid Advisory Board member for F. Hoffmann-La Roche Ltd and uniQure biopharma B.V., and he is a paid consultant for Vaccinex Inc, Wave Life Sciences USA Inc, Genentech Inc, and Triplet Therapeutics Inc.
J.F.G. is a Scientific Advisory Board member and has a financial interest in Triplet Therapeutics, Inc. His NIH-funded project is using genetic and genomic approaches to uncover other genes that significantly influence when diagnosable symptoms emerge and how rapidly they worsen in Huntington Disease. The company is developing new therapeutic approaches to address triplet repeat disorders such Huntington’s Disease, Myotonic Dystrophy and spinocerebellar ataxias. His interests were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies.
Figures
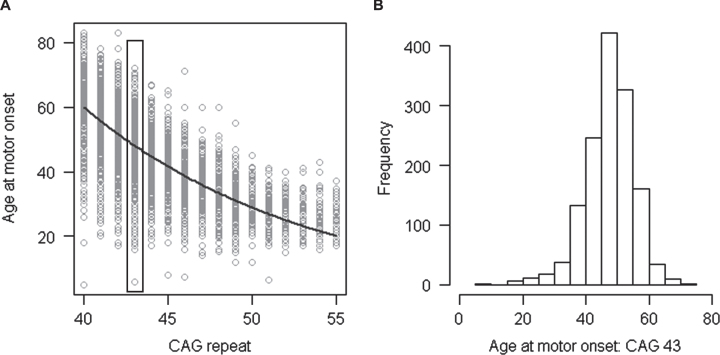
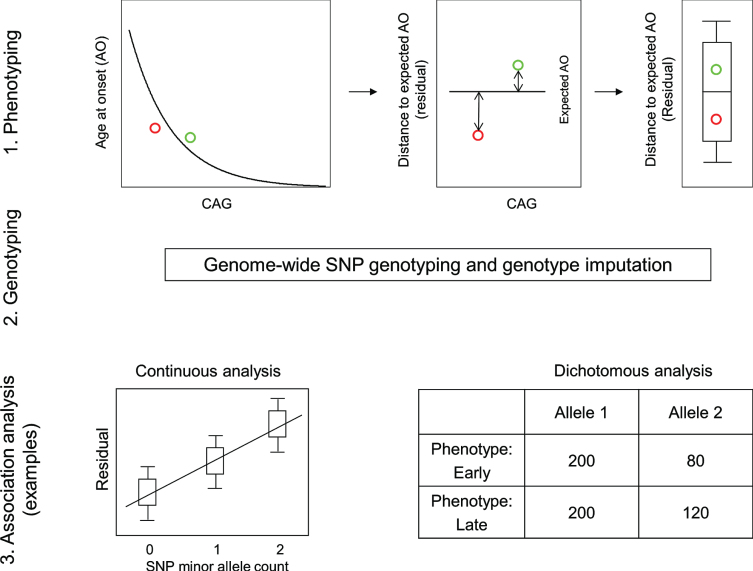
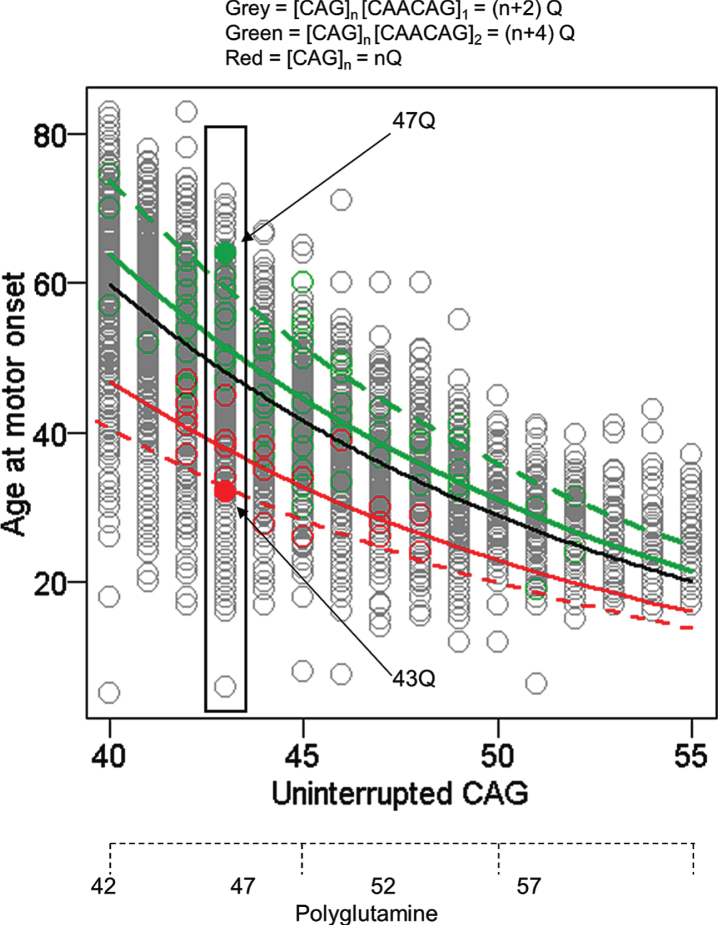
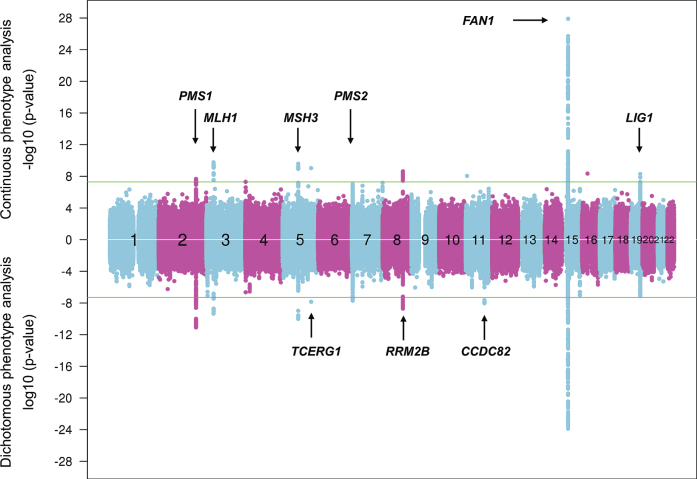

References
-
- Gusella JF, Wexler NS, Conneally PM, Naylor SL, Anderson MA, Tanzi RE, et al.. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature. 1983;306(5940):234–8. - PubMed
-
- Meissen GJ, Myers RH, Mastromauro CA, Koroshetz WJ, Klinger KW, Farrer LA, et al.. Predictive testing for Huntington’s disease with use of a linked DNA marker. N Engl J Med. 1988;318(9):535–42. - PubMed
-
- Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72(6):971–83. - PubMed
-
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352(6330):77–9. - PubMed
-
- Kremer EJ, Pritchard M, Lynch M, Yu S, Holman K, Baker E, et al.. Mapping of DNA instability at the fragile X to a trinucleotide repeat sequence p(CCG)n. Science. 1991;252(5013):1711–4. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
