

Approaches to Sequence the HTT CAG Repeat Expansion and Quantify Repeat Length Variation
- PMID: 33579864
- PMCID: PMC7990409
- DOI: 10.3233/JHD-200433
Approaches to Sequence the HTT CAG Repeat Expansion and Quantify Repeat Length Variation
Abstract
Background: Huntington's disease (HD) is an autosomal dominant neurodegenerative disorder caused by the expansion of the HTT CAG repeat. Affected individuals inherit ≥36 repeats and longer alleles cause earlier onset, greater disease severity and faster disease progression. The HTT CAG repeat is genetically unstable in the soma in a process that preferentially generates somatic expansions, the proportion of which is associated with disease onset, severity and progression. Somatic mosaicism of the HTT CAG repeat has traditionally been assessed by semi-quantitative PCR-electrophoresis approaches that have limitations (e.g., no information about sequence variants). Genotyping-by-sequencing could allow for some of these limitations to be overcome.
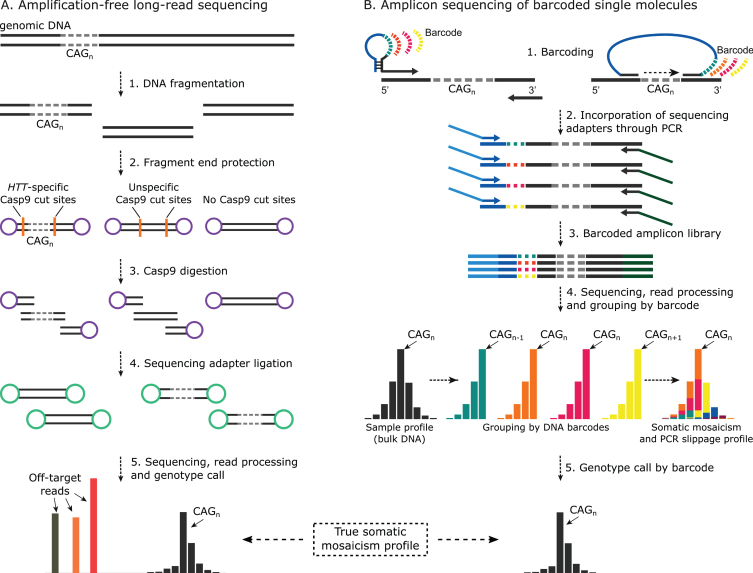
Objective: To investigate the utility of PCR sequencing to genotype large (>50 CAGs) HD alleles and to quantify the associated somatic mosaicism.
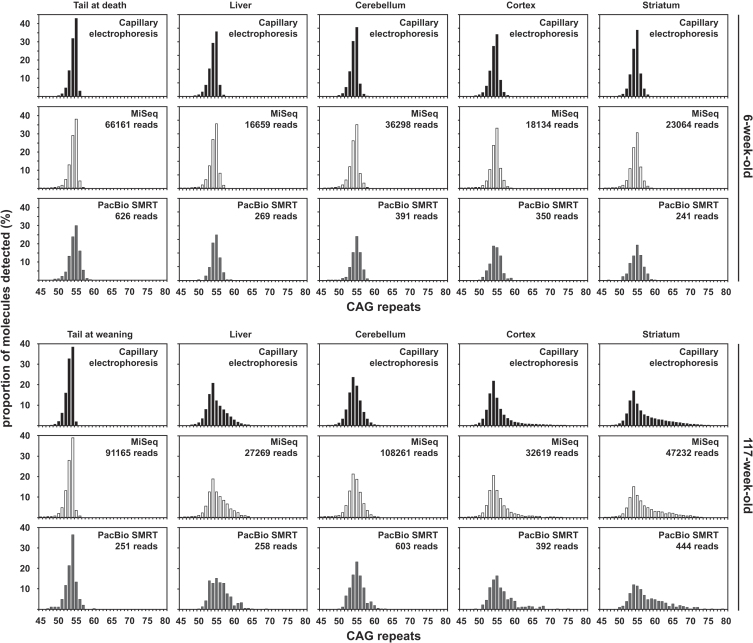
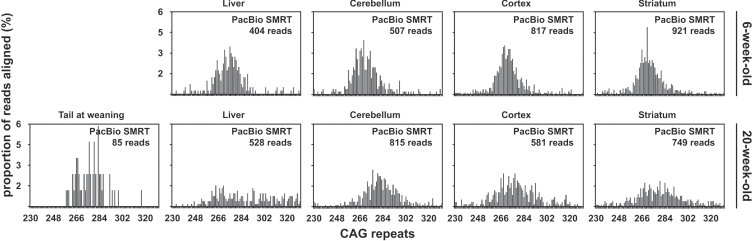
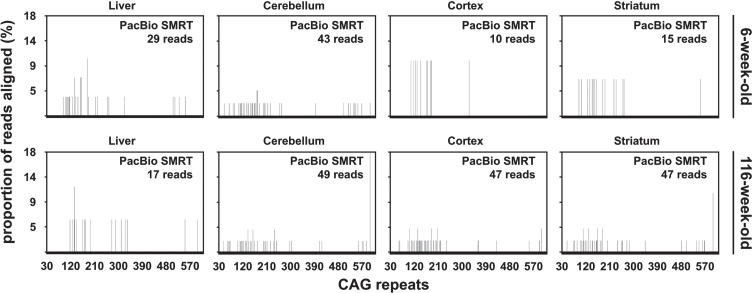
Methods: We have applied MiSeq and PacBio sequencing to PCR products of the HTT CAG repeat in transgenic R6/2 mice carrying ∼55, ∼110, ∼255 and ∼470 CAGs. For each of these alleles, we compared the repeat length distributions generated for different tissues at two ages.
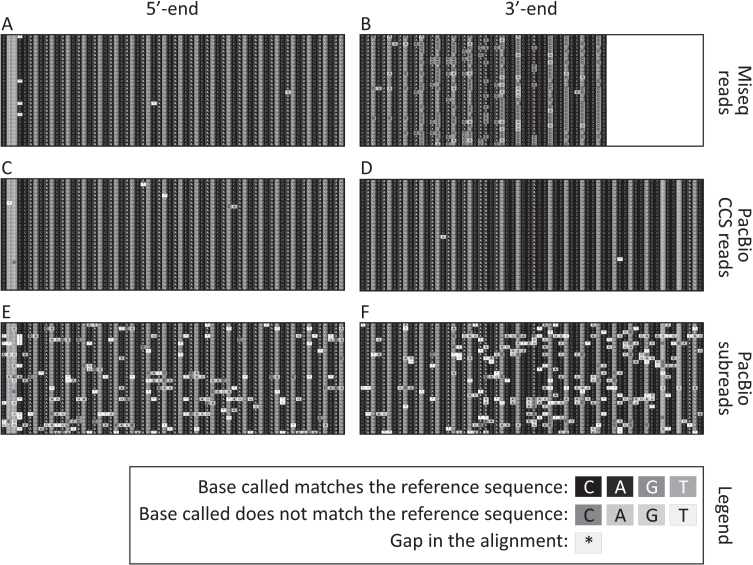
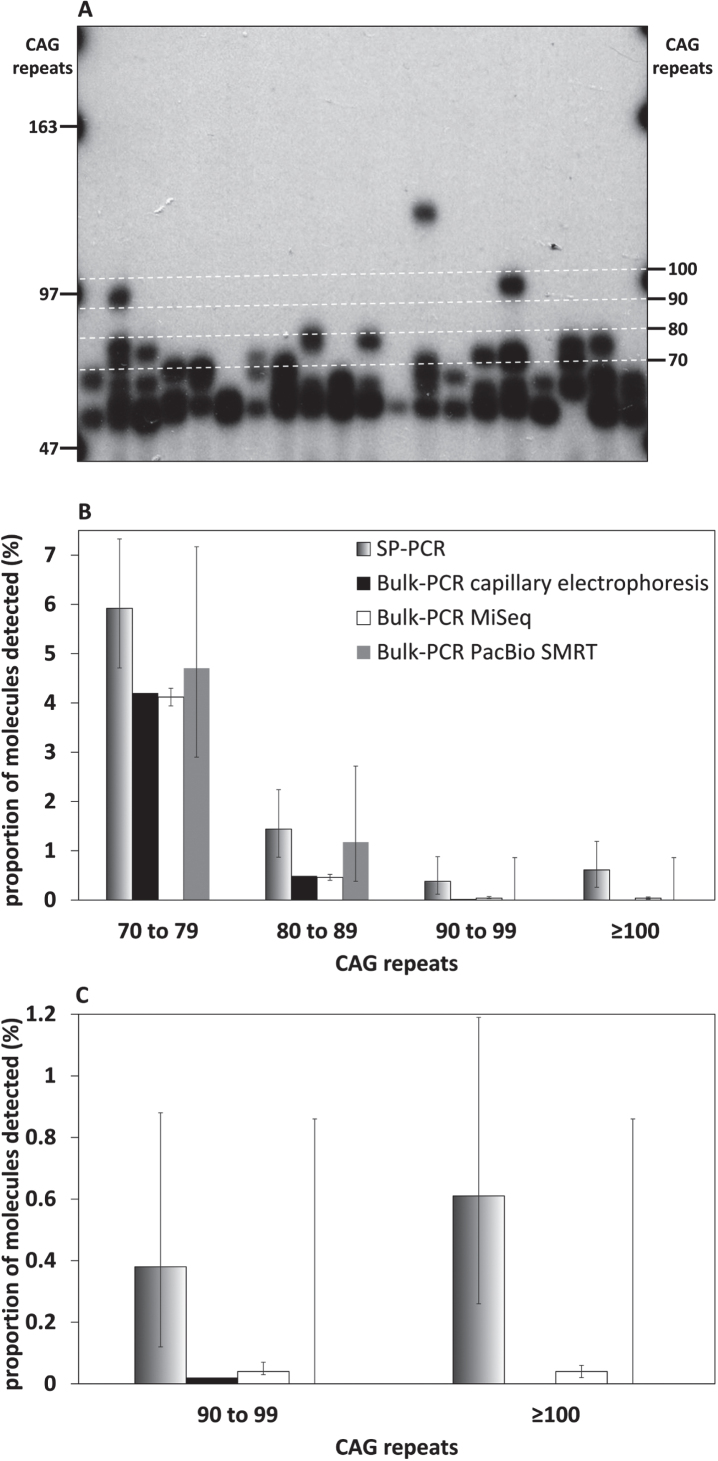
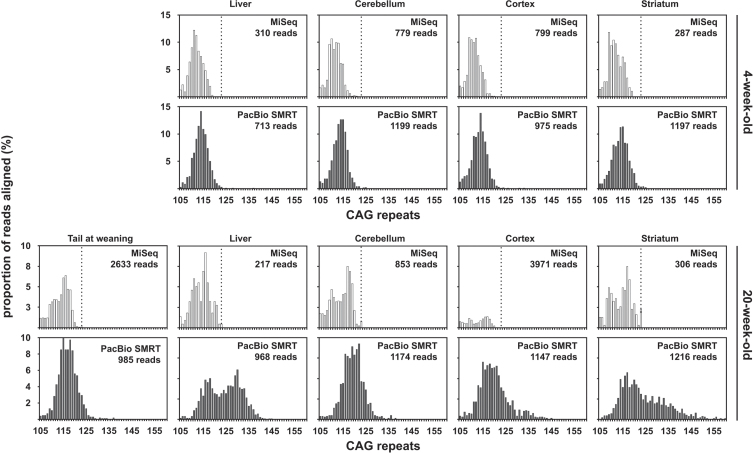
Results: We were able to sequence the CAG repeat full length in all samples. However, the repeat length distributions for samples with ∼470 CAGs were biased towards shorter repeat lengths.
Conclusion: PCR sequencing can be used to sequence all the HD alleles considered, but this approach cannot be used to estimate modal allele size or quantify somatic expansions for alleles ⪢250 CAGs. We review the limitations of PCR sequencing and alternative approaches that may allow the quantification of somatic contractions and very large somatic expansions.
Keywords: Huntington disease; Somatic mosaicism; huntingtin; parallel sequencing; repeat expansion.
Conflict of interest statement
V.C.W. is a scientific advisory board member of Triplet Therapeutics, a company developing new therapeutic approaches to address triplet repeat disorders such as HD and myotonic dystrophy and of LoQus23 Therapeutics, and has provided paid consulting services to Alnylam. Her financial interests in Triplet Therapeutics were reviewed and are managed by Massachusetts General Hospital and Partners HealthCare in accordance with their conflict of interest policies. S.K. is employed by CHDI Management, Inc., as an advisor to the CHDI Foundation. D.G.M. has been a scientific consultant and/or received honoraria or stock options from Biogen Idec, AMO Pharma, Charles River, Vertex Pharmaceuticals, Triplet Therapeutics, LoQus23, and Small Molecule RNA and has had research contracts with AMO Pharma and Vertex Pharmaceuticals.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
