

Current Therapeutic Approaches in FSHD
- PMID: 33579868
- PMCID: PMC8203219
- DOI: 10.3233/JND-200554
Current Therapeutic Approaches in FSHD
Abstract
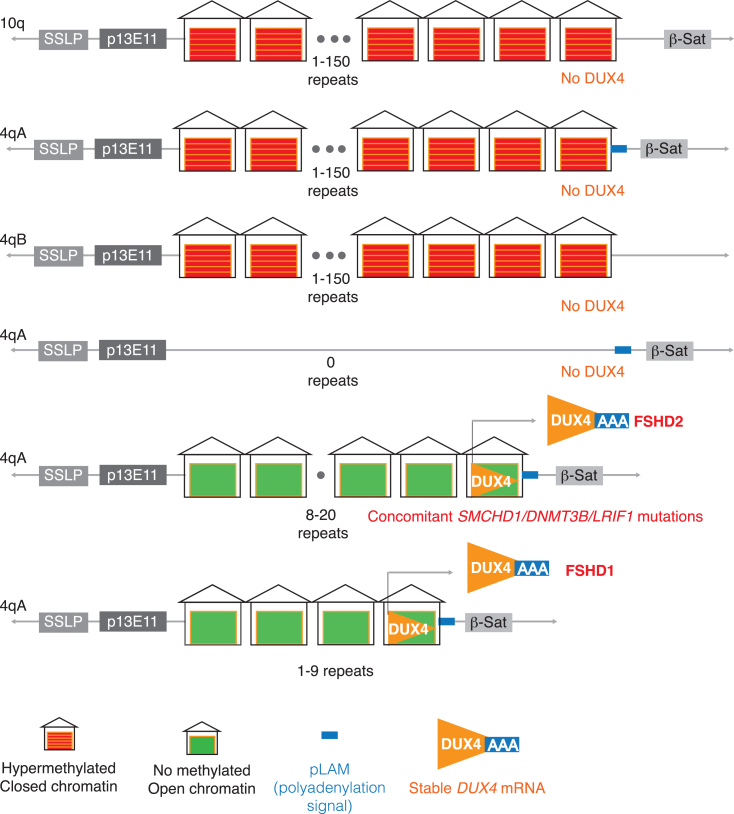
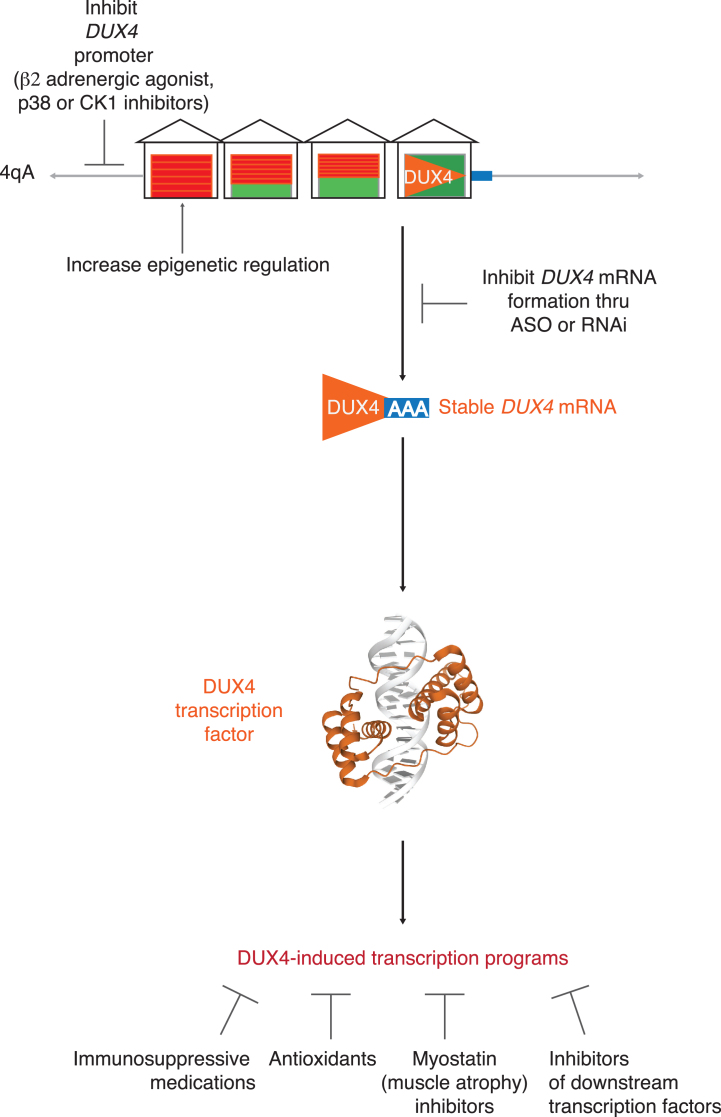
Facioscapulohumeral muscular dystrophy (FSHD) is one of the most common muscular dystrophies. Over the last decade, a consensus was reached regarding the underlying cause of FSHD allowing-for the first time-a targeted approach to treatment. FSHD is the result of a toxic gain-of-function from de-repression of the DUX4 gene, a gene not normally expressed in skeletal muscle. With a clear therapeutic target, there is increasing interest in drug development for FSHD, an interest buoyed by the recent therapeutic successes in other neuromuscular diseases. Herein, we review the underlying disease mechanism, potential therapeutic approaches as well as the state of trial readiness in the planning and execution of future clinical trials in FSHD.
Keywords: All neuromuscular disease; facioscapulohumeral dystrophy (FSHD); muscle disease; outcome measures.
Conflict of interest statement
Dr. Wang reports consultancy for Biogen. Dr. Tawil reports consultancy with Fulcrum Therapeutics and Acceleron Pharma.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
