

AA amyloid fibrils from diseased tissue are structurally different from in vitro formed SAA fibrils
- PMID: 33579941
- PMCID: PMC7881110
- DOI: 10.1038/s41467-021-21129-z
AA amyloid fibrils from diseased tissue are structurally different from in vitro formed SAA fibrils
Abstract
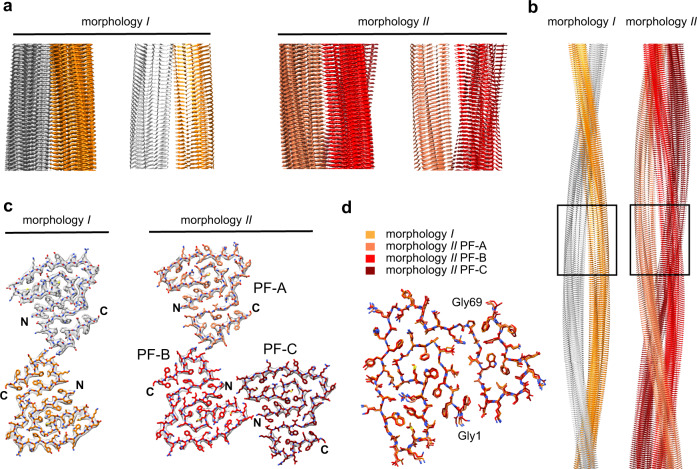
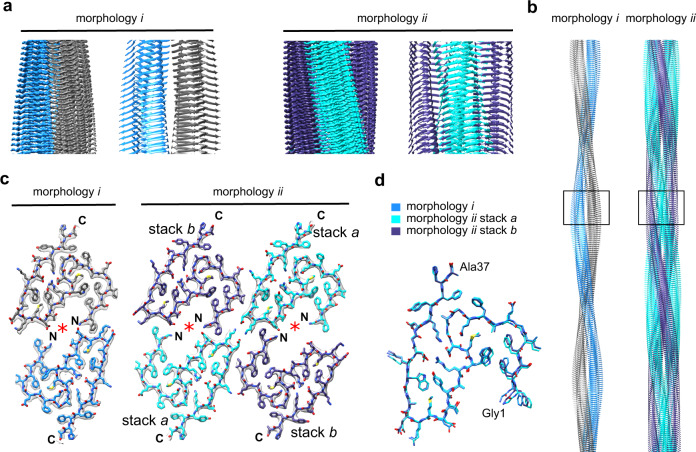
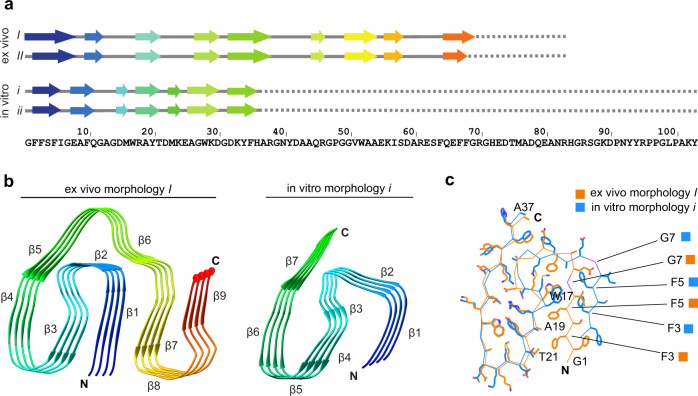
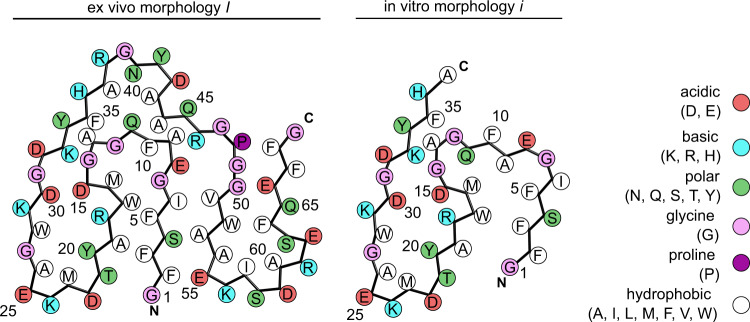
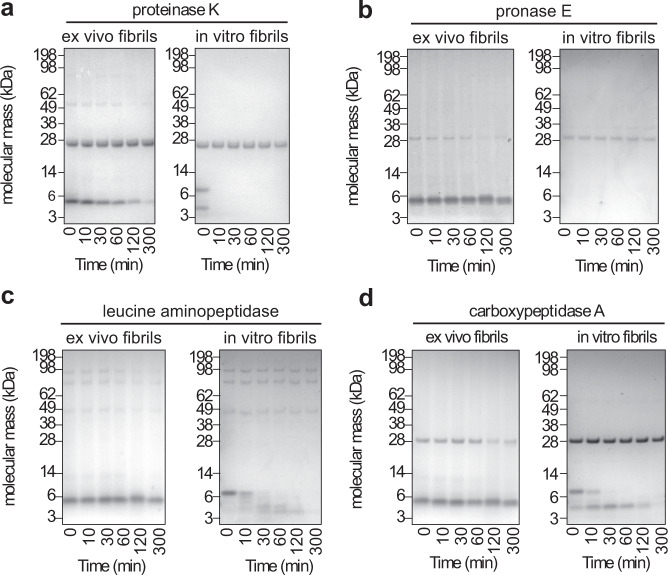
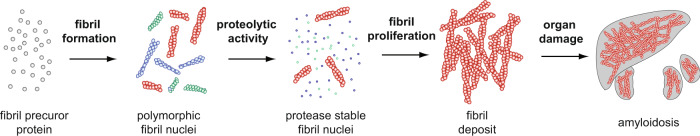
Systemic AA amyloidosis is a world-wide occurring protein misfolding disease of humans and animals. It arises from the formation of amyloid fibrils from serum amyloid A (SAA) protein. Using cryo electron microscopy we here show that amyloid fibrils which were purified from AA amyloidotic mice are structurally different from fibrils formed from recombinant SAA protein in vitro. Ex vivo amyloid fibrils consist of fibril proteins that contain more residues within their ordered parts and possess a higher β-sheet content than in vitro fibril proteins. They are also more resistant to proteolysis than their in vitro formed counterparts. These data suggest that pathogenic amyloid fibrils may originate from proteolytic selection, allowing specific fibril morphologies to proliferate and to cause damage to the surrounding tissue.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
