

Commonalities across computational workflows for uncovering explanatory variants in undiagnosed cases
- PMID: 33580225
- PMCID: PMC8187147
- DOI: 10.1038/s41436-020-01084-8
Commonalities across computational workflows for uncovering explanatory variants in undiagnosed cases
Abstract
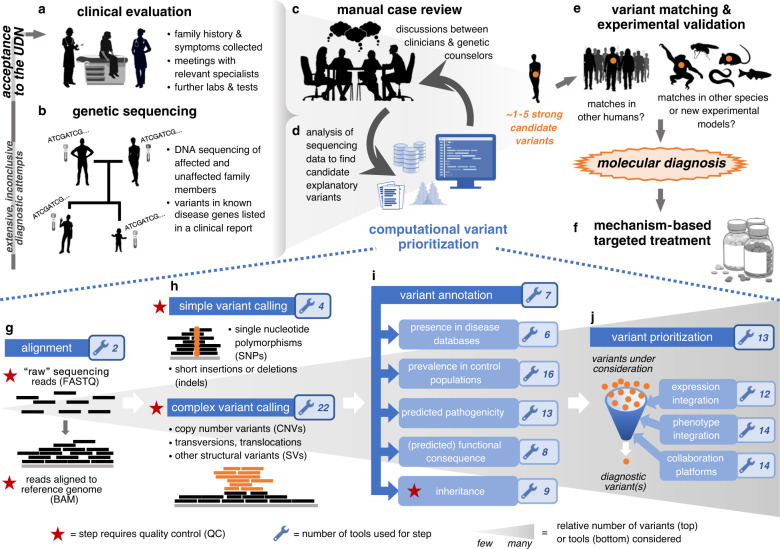
Purpose: Genomic sequencing has become an increasingly powerful and relevant tool to be leveraged for the discovery of genetic aberrations underlying rare, Mendelian conditions. Although the computational tools incorporated into diagnostic workflows for this task are continually evolving and improving, we nevertheless sought to investigate commonalities across sequencing processing workflows to reveal consensus and standard practice tools and highlight exploratory analyses where technical and theoretical method improvements would be most impactful.
Methods: We collected details regarding the computational approaches used by a genetic testing laboratory and 11 clinical research sites in the United States participating in the Undiagnosed Diseases Network via meetings with bioinformaticians, online survey forms, and analyses of internal protocols.
Results: We found that tools for processing genomic sequencing data can be grouped into four distinct categories. Whereas well-established practices exist for initial variant calling and quality control steps, there is substantial divergence across sites in later stages for variant prioritization and multimodal data integration, demonstrating a diversity of approaches for solving the most mysterious undiagnosed cases.
Conclusion: The largest differences across diagnostic workflows suggest that advances in structural variant detection, noncoding variant interpretation, and integration of additional biomedical data may be especially promising for solving chronically undiagnosed cases.
Conflict of interest statement
P.L. is an employee of Baylor College of Medicine and derives support through a professional services agreement with Baylor Genetics, which performs clinical genetic testing services. The other authors declare no competing interests.
Figures
References
-
- Online Mendelian Inheritance in Man, OMIM. (McKusick-Nathans Institute of Genetic Medicine, Johns Hopkins University, Baltimore, MD). https://omim.org.
Publication types
MeSH terms
Grants and funding
- U01 HG007690/HG/NHGRI NIH HHS/United States
- U01 NS134353/NS/NINDS NIH HHS/United States
- U01 HG010215/HG/NHGRI NIH HHS/United States
- U01 HG007674/HG/NHGRI NIH HHS/United States
- U01 HG007672/HG/NHGRI NIH HHS/United States
- U01 HG010218/HG/NHGRI NIH HHS/United States
- U01 HG010233/HG/NHGRI NIH HHS/United States
- U01 HG010230/HG/NHGRI NIH HHS/United States
- U01 HG010217/HG/NHGRI NIH HHS/United States
- U01 HG007530/HG/NHGRI NIH HHS/United States
- U01 HG007703/HG/NHGRI NIH HHS/United States
- U01 HG007942/HG/NHGRI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
