

High-resolution quantitative profiling of tRNA abundance and modification status in eukaryotes by mim-tRNAseq
- PMID: 33581077
- PMCID: PMC8062790
- DOI: 10.1016/j.molcel.2021.01.028
High-resolution quantitative profiling of tRNA abundance and modification status in eukaryotes by mim-tRNAseq
Abstract
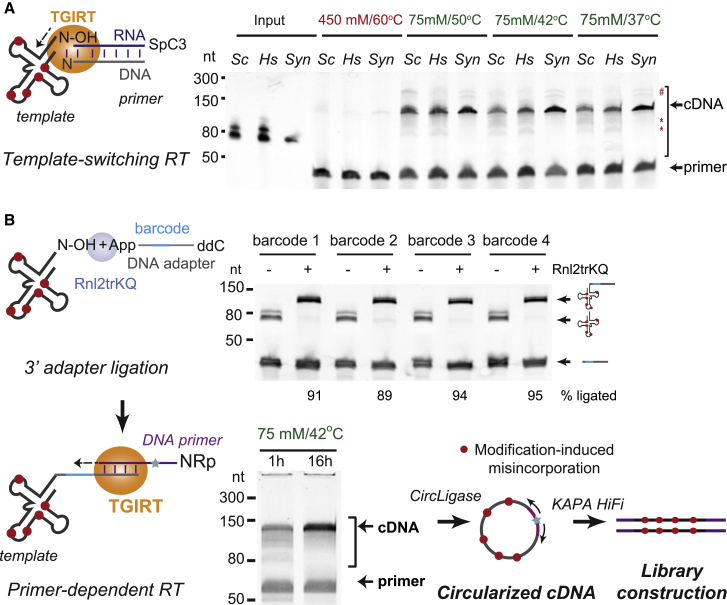
Measurements of cellular tRNA abundance are hampered by pervasive blocks to cDNA synthesis at modified nucleosides and the extensive similarity among tRNA genes. We overcome these limitations with modification-induced misincorporation tRNA sequencing (mim-tRNAseq), which combines a workflow for full-length cDNA library construction from endogenously modified tRNA with a comprehensive and user-friendly computational analysis toolkit. Our method accurately captures tRNA abundance and modification status in yeast, fly, and human cells and is applicable to any organism with a known genome. We applied mim-tRNAseq to discover a dramatic heterogeneity of tRNA isodecoder pools among diverse human cell lines and a surprising interdependence of modifications at distinct sites within the same tRNA transcript.
Keywords: RNA modification; tRNA quantitation; transfer RNA; translation regulation.
Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures






Comment in
-
How many tRNAs are out there?Mol Cell. 2021 Apr 15;81(8):1595-1597. doi: 10.1016/j.molcel.2021.03.021. Mol Cell. 2021. PMID: 33861948
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
