

Enhanced antitumor efficacy of a novel oncolytic vaccinia virus encoding a fully monoclonal antibody against T-cell immunoglobulin and ITIM domain (TIGIT)
- PMID: 33581644
- PMCID: PMC7878184
- DOI: 10.1016/j.ebiom.2021.103240
Enhanced antitumor efficacy of a novel oncolytic vaccinia virus encoding a fully monoclonal antibody against T-cell immunoglobulin and ITIM domain (TIGIT)
Abstract
Background: Oncolytic virotherapy with vaccinia virus (VV) can lead to effective anti-tumor immunity by turning "cold" tumors into "hot" tumors. However, its therapeutic potential is affected by the tumor's local immunosuppressive tumor microenvironment (TME). Therefore, it is necessary to explore the use of immune checkpoint inhibitors to arm oncolytic VVs to enhance their anti-tumor efficacy.
Methods: A novel recombinant oncolytic VV, VV-α-TIGIT, which encoded a fully monoclonal antibody against T-cell immunoglobulin and ITIM domain (TIGIT) was generated by homologous recombination with a shuttle plasmid. The anti-tumor efficacy of the VV-α-TIGIT was investigated in several subcutaneous and ascites tumor models.
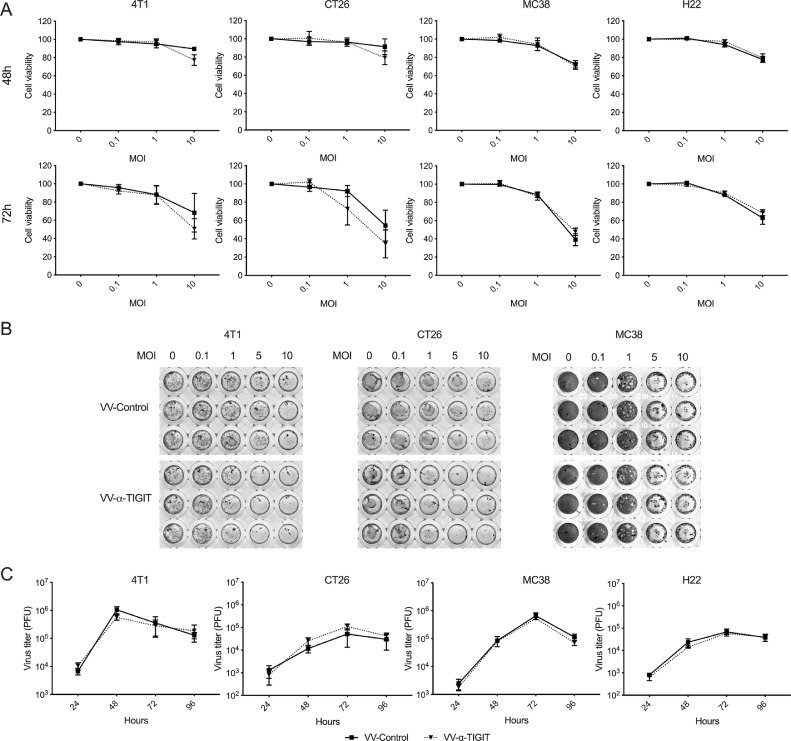
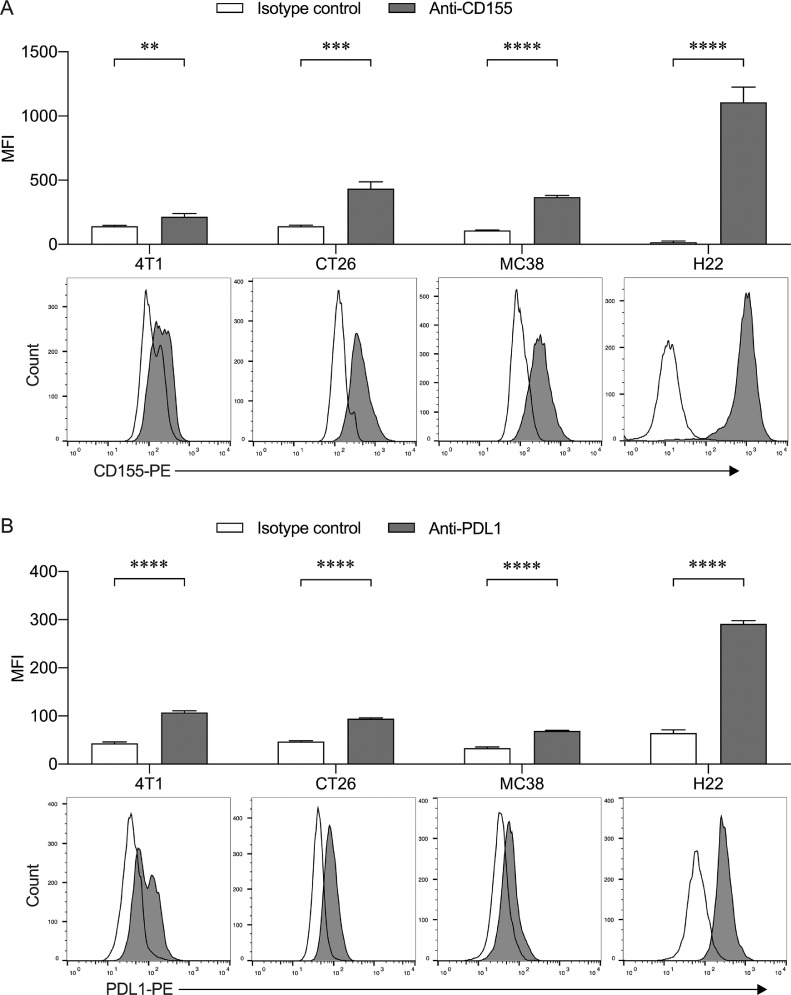
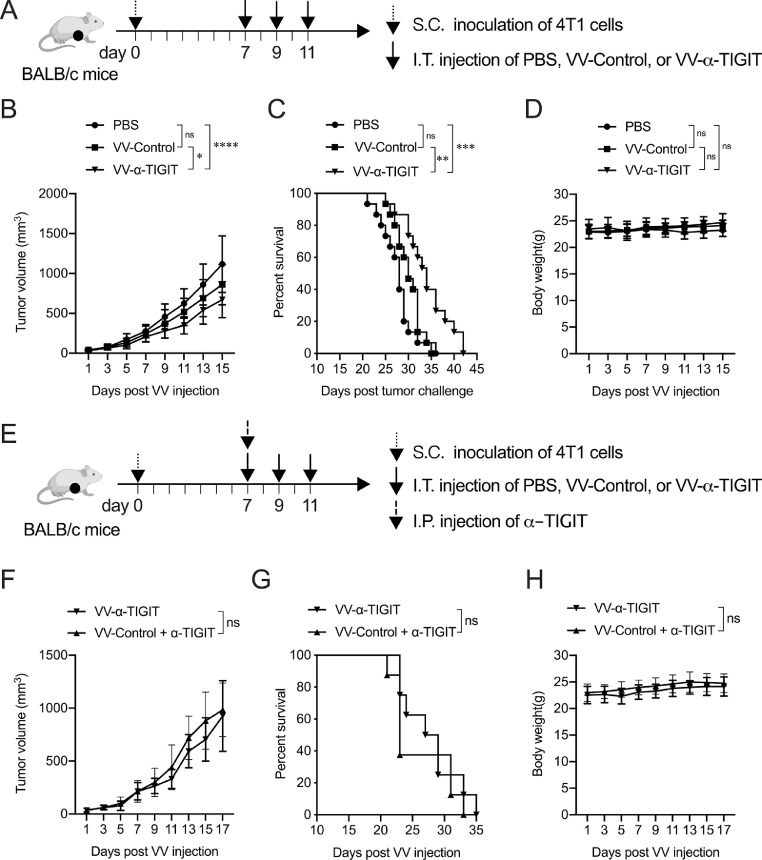
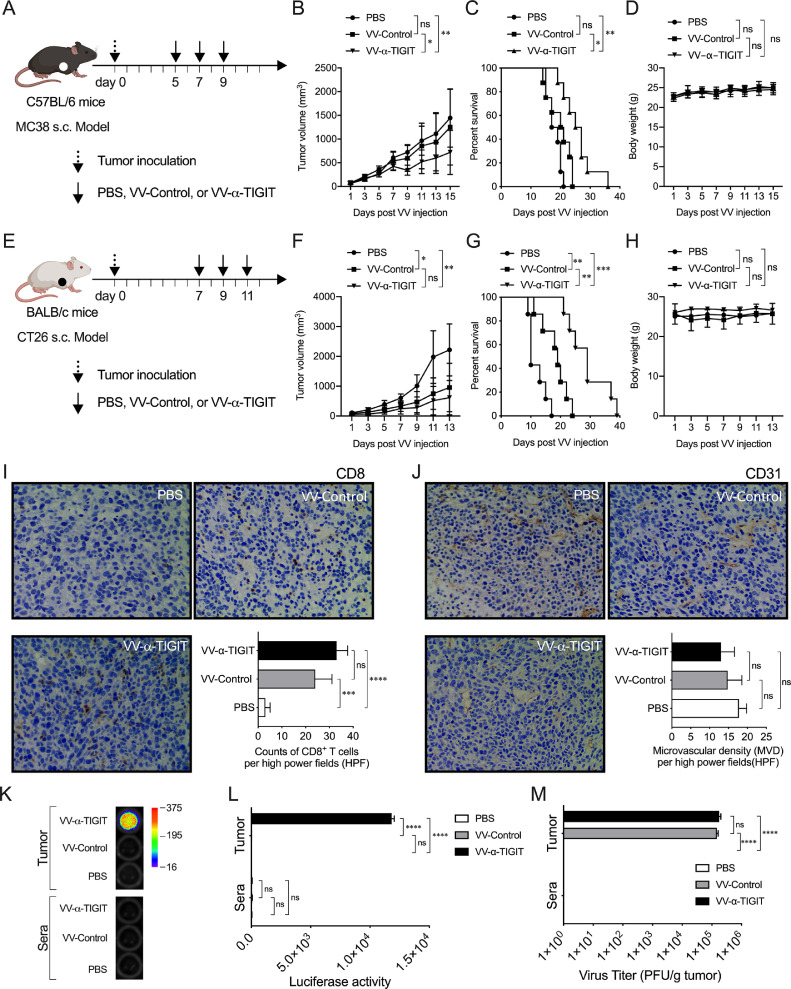
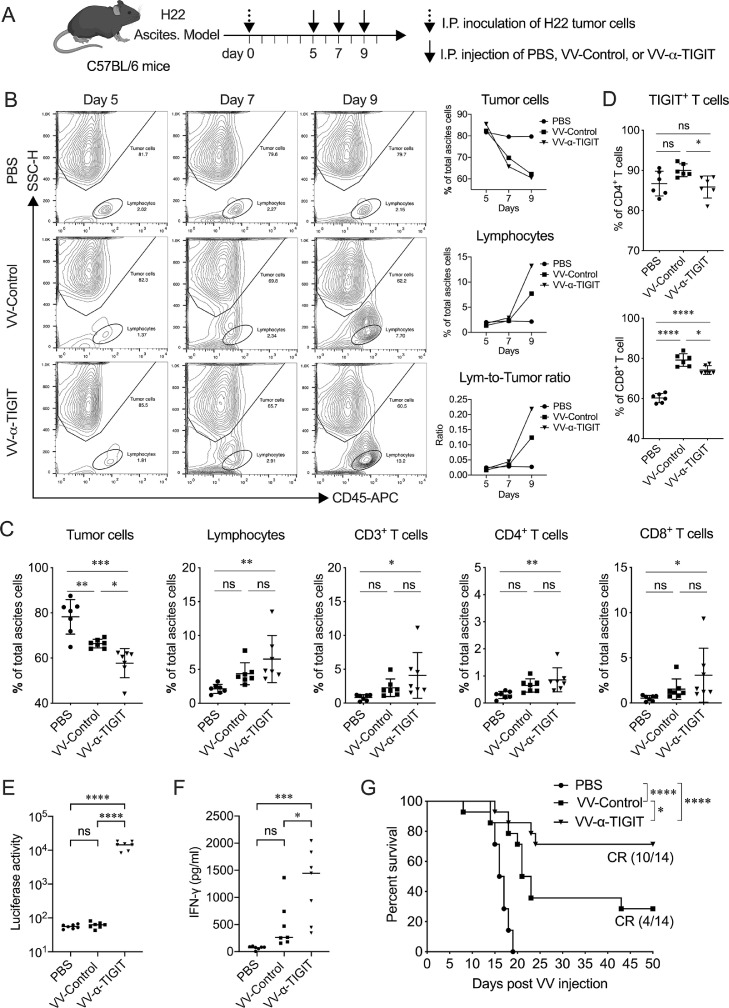
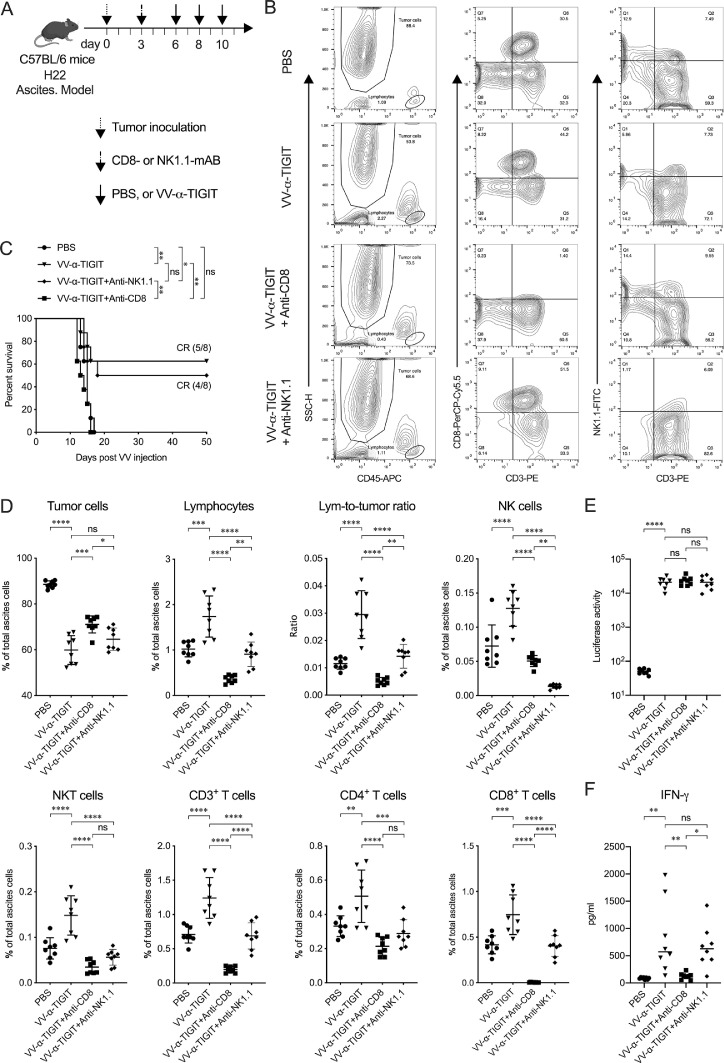
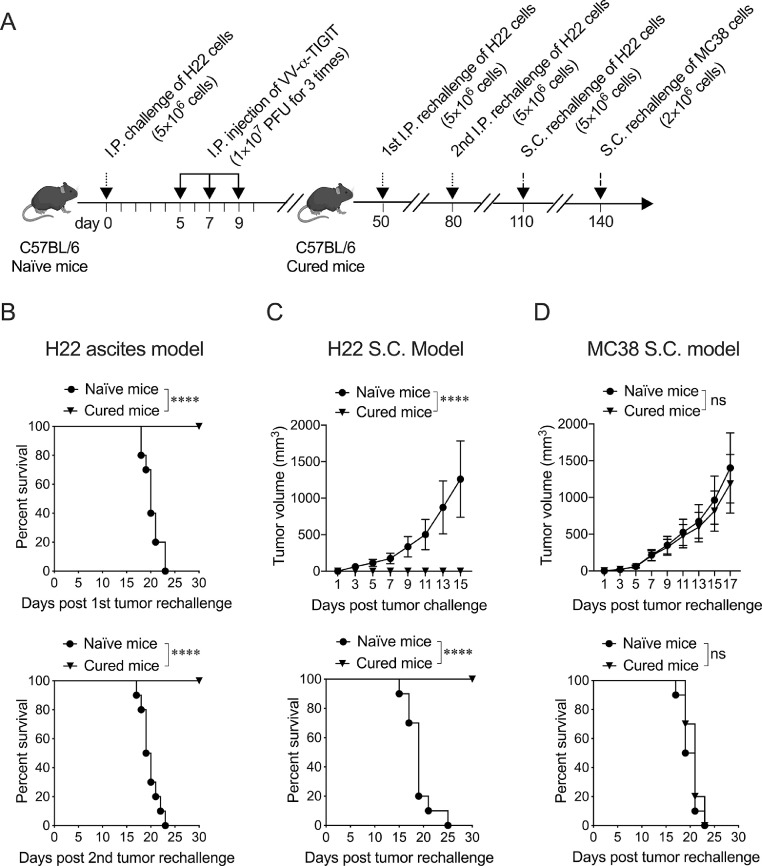
Findings: The functional α-TIGIT was sufficiently produced and secreted by tumor cells infected with VV-α-TIGIT, which effectively replicated in tumor cells leading to significant oncolysis. Intratumoral injection of VV-α-TIGIT improved anti-tumor efficacy in several murine subcutaneous tumor models compared to VV-Control (without α-TIGIT insertion). Intraperitoneal injection of VV-α-TIGIT achieved approximately 70% of complete tumor regression in an ascites tumor model. At the same time, treatment with VV-α-TIGIT significantly increased the recruitment and activation of T cells in TME. Moreover, the in vivo anti-tumor activity of VV-α-TIGIT was largely dependent on CD8+ T cell-mediated immunity. Finally, the tumor-bearing mice cured of VV-α-TIGIT treatment resisted rechallenge with the same tumor cells, suggesting a long-term persistence of tumor-specific immunological memory.
Interpretation: The recombinant oncolytic virus VV-α-TIGIT successfully combines the advantages of oncolytic virotherapy and intratumorally expression of immune checkpoint inhibitor against TIGIT. This novel strategy can provide information on the optimal design of novel antibody-armed oncolytic viruses for cancer immunotherapy.
Funding: This work was supported by the National Natural Science Foundation of China (81773255, 81472820, and 81700037), the Science and Technology Innovation Foundation of Nanjing University (14913414), and the Natural Science Foundation of Jiangsu Province of China (BK20171098).
Keywords: Cancer immunotherapy; Checkpoint inhibitors; Oncolytic virus; TIGIT; Vaccinia virus.
Copyright © 2021 The Authors. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of Interests The authors disclose no conflicts of interest.
Figures

References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
