

Interleukin-1 as Innate Mediator of T Cell Immunity
- PMID: 33584721
- PMCID: PMC7873566
- DOI: 10.3389/fimmu.2020.621931
Interleukin-1 as Innate Mediator of T Cell Immunity
Abstract
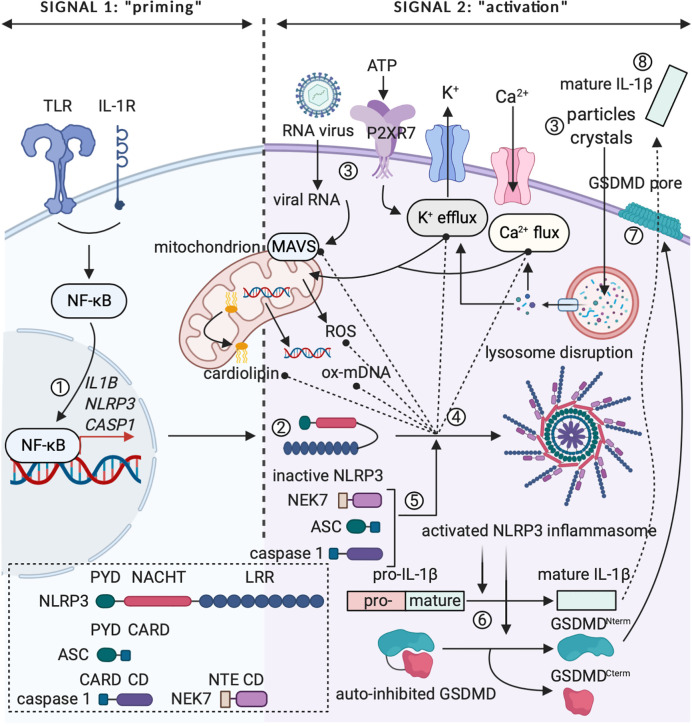
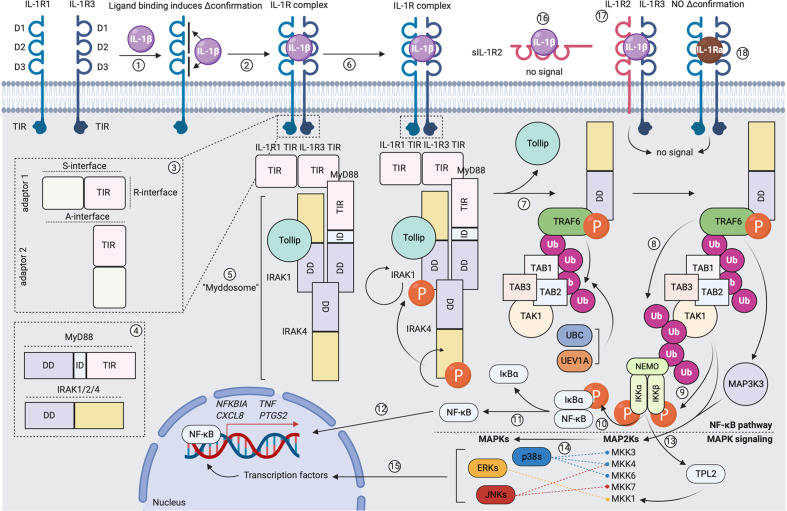
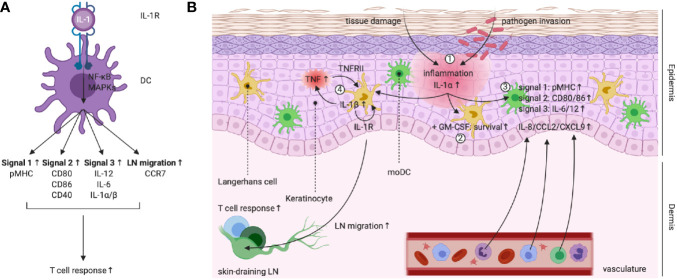
The three-signal paradigm tries to capture how the innate immune system instructs adaptive immune responses in three well-defined actions: (1) presentation of antigenic peptides in the context of MHC molecules, which allows for a specific T cell response; (2) T cell co-stimulation, which breaks T cell tolerance; and (3) secretion of polarizing cytokines in the priming environment, thereby specializing T cell immunity. The three-signal model provides an empirical framework for innate instruction of adaptive immunity, but mainly discusses STAT-dependent cytokines in T cell activation and differentiation, while the multi-faceted roles of type I IFNs and IL-1 cytokine superfamily members are often neglected. IL-1α and IL-1β are pro-inflammatory cytokines, produced following damage to the host (release of DAMPs) or upon innate recognition of PAMPs. IL-1 activity on both DCs and T cells can further shape the adaptive immune response with variable outcomes. IL-1 signaling in DCs promotes their ability to induce T cell activation, but also direct action of IL-1 on both CD4+ and CD8+ T cells, either alone or in synergy with prototypical polarizing cytokines, influences T cell differentiation under different conditions. The activities of IL-1 form a direct bridge between innate and adaptive immunity and could therefore be clinically translatable in the context of prophylactic and therapeutic strategies to empower the formation of T cell immunity. Understanding the modalities of IL-1 activity during T cell activation thus could hold major implications for rational development of the next generation of vaccine adjuvants.
Keywords: CD4+ T cells; CD8+ T cells; cancer immunotherapy; cellular adjuvant; dendritic cells; interleukin-1; vaccination.
Copyright © 2021 Van Den Eeckhout, Tavernier and Gerlo.
Conflict of interest statement
JT is affiliated with Orionis Biosciences BV as scientific advisor and holds equity interests in Orionis Biosciences BV. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures



References
-
- Janeway CA. a. Pillars article: approaching the asymptote? Evolution and revolution in immunology. Cold spring harb symp quant biol. 1989. 54: 1-13. J Immunol (2013) 191:4475–87. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials