

Characteristics of Genetic Variations Associated With Lennox-Gastaut Syndrome in Korean Families
- PMID: 33584793
- PMCID: PMC7874053
- DOI: 10.3389/fgene.2020.590924
Characteristics of Genetic Variations Associated With Lennox-Gastaut Syndrome in Korean Families
Erratum in
-
Corrigendum: Characteristics of Genetic Variations Associated With Lennox-Gastaut Syndrome in Korean Families.Front Genet. 2021 Mar 5;12:669107. doi: 10.3389/fgene.2021.669107. eCollection 2021. Front Genet. 2021. PMID: 33747056 Free PMC article.
Abstract
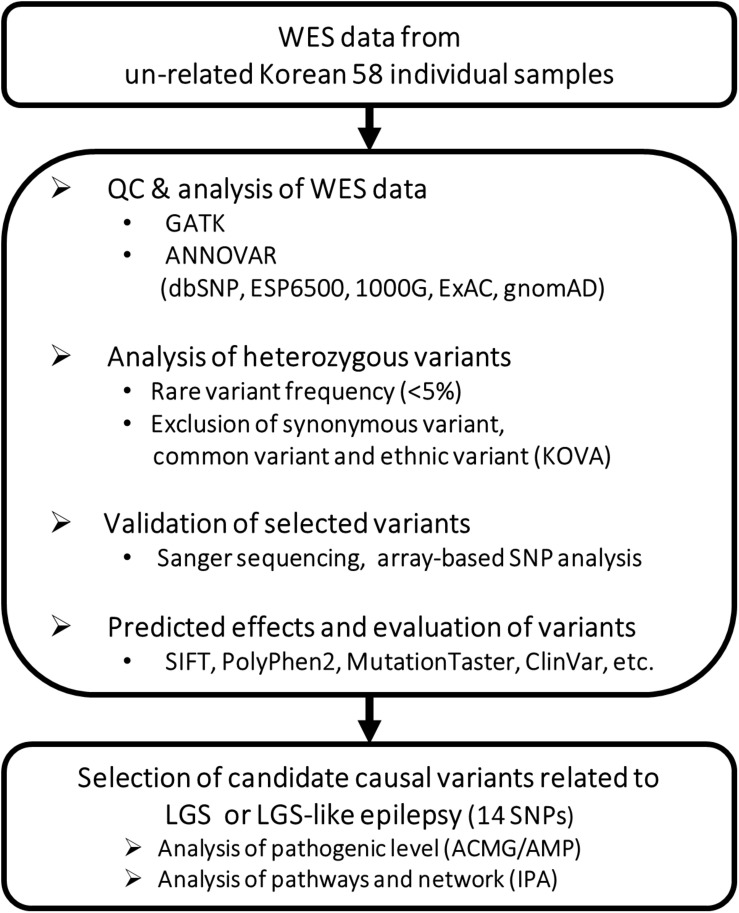
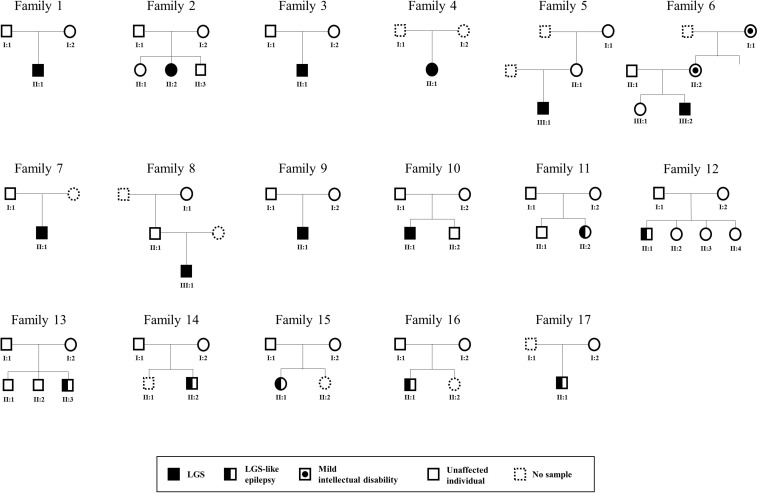
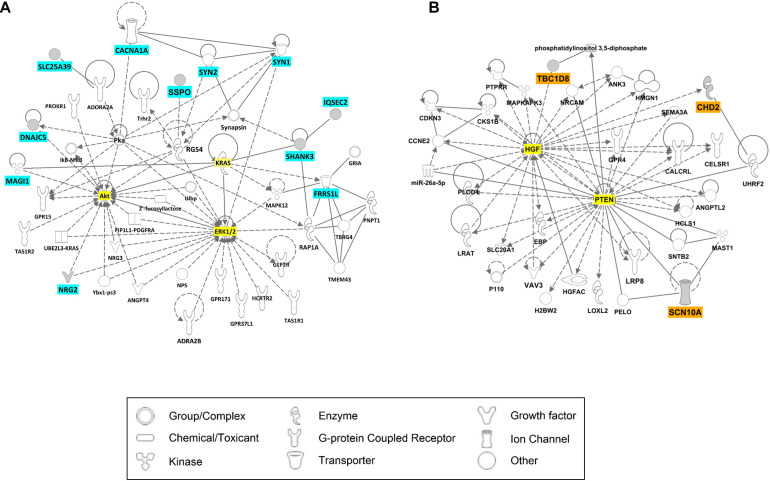
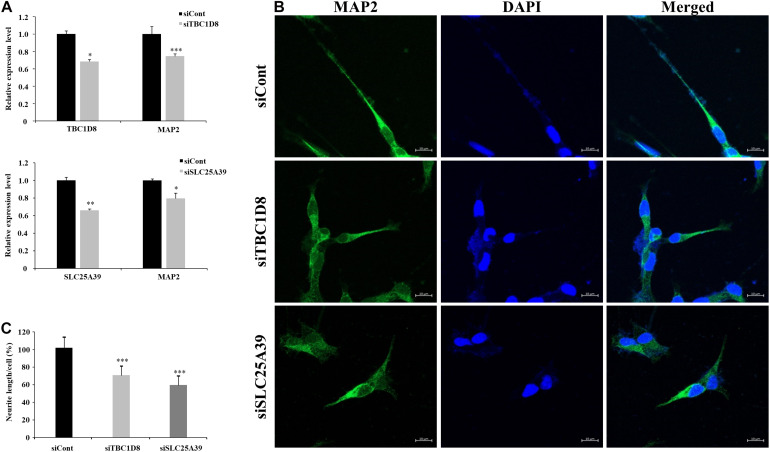
Lennox-Gastaut syndrome (LGS) is a severe type of childhood-onset epilepsy characterized by multiple types of seizures, specific discharges on electroencephalography, and intellectual disability. Most patients with LGS do not respond well to drug treatment and show poor long-term prognosis. Approximately 30% of patients without brain abnormalities have unidentifiable causes. Therefore, accurate diagnosis and treatment of LGS remain challenging. To identify causative mutations of LGS, we analyzed the whole-exome sequencing data of 17 unrelated Korean families, including patients with LGS and LGS-like epilepsy without brain abnormalities, using the Genome Analysis Toolkit. We identified 14 mutations in 14 genes as causes of LGS or LGS-like epilepsy. 64 percent of the identified genes were reported as LGS or epilepsy-related genes. Many of these variations were novel and considered as pathogenic or likely pathogenic. Network analysis was performed to classify the identified genes into two network clusters: neuronal signal transmission or neuronal development. Additionally, knockdown of two candidate genes with insufficient evidence of neuronal functions, SLC25A39 and TBC1D8, decreased neurite outgrowth and the expression level of MAP2, a neuronal marker. These results expand the spectrum of genetic variations and may aid the diagnosis and management of individuals with LGS.
Keywords: Lennox-Gastaut syndrome; Rare-diseases; epilepsy; genetic variation; whole-exome sequencing.
Copyright © 2021 Yang, Choi, Yoon, Lee, Nam, Jun, Kwon, Yun, Jeon, Byeon, Halder, Kong, Lee, Lee, Kang and Kim.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

References
LinkOut - more resources
Full Text Sources
Other Literature Sources