

Severe insulin resistance syndromes
- PMID: 33586681
- PMCID: PMC7880309
- DOI: 10.1172/JCI142245
Severe insulin resistance syndromes
Abstract
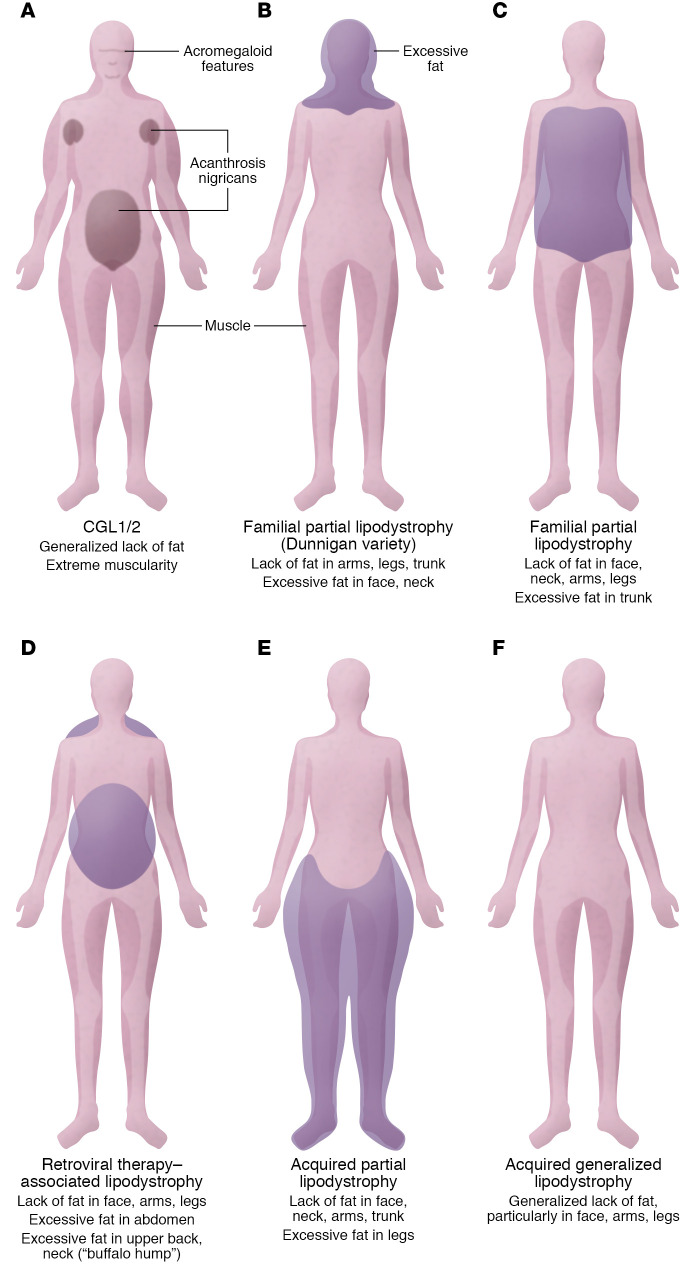
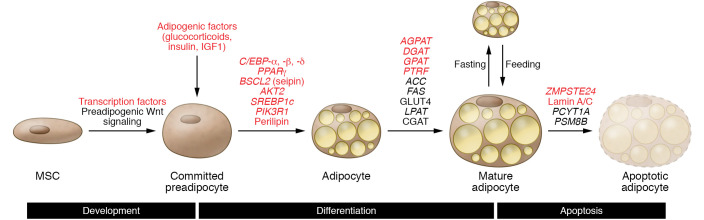
Severe insulin resistance syndromes are a heterogeneous group of rare disorders characterized by profound insulin resistance, substantial metabolic abnormalities, and a variety of clinical manifestations and complications. The etiology of these syndromes may be hereditary or acquired, due to defects in insulin potency and action, cellular responsiveness to insulin, and/or aberrations in adipose tissue function or development. Over the past decades, advances in medical technology, particularly in genomic technologies and genetic analyses, have provided insights into the underlying pathophysiological pathways and facilitated the more precise identification of several of these conditions. However, the exact cellular and molecular mechanisms of insulin resistance have not yet been fully elucidated for all syndromes. Moreover, in clinical practice, many of the syndromes are often misdiagnosed or underdiagnosed. The majority of these disorders associate with an increased risk of severe complications and mortality; thus, early identification and personalized clinical management are of the essence. This Review aims to categorize severe insulin resistance syndromes by disease process, including insulin receptor defects, signaling defects, and lipodystrophies. We also highlight several complex syndromes and emphasize the need to identify patients, investigate underlying disease mechanisms, and develop specific treatment regimens.
Conflict of interest statement
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
