

Of Traits and Trees: Probabilistic Distances under Continuous Trait Models for Dissecting the Interplay among Phylogeny, Model, and Data
- PMID: 33587145
- PMCID: PMC8208806
- DOI: 10.1093/sysbio/syab009
Of Traits and Trees: Probabilistic Distances under Continuous Trait Models for Dissecting the Interplay among Phylogeny, Model, and Data
Abstract
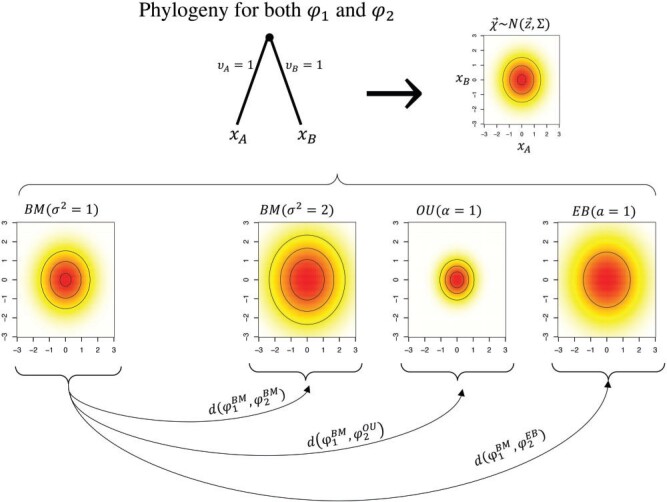
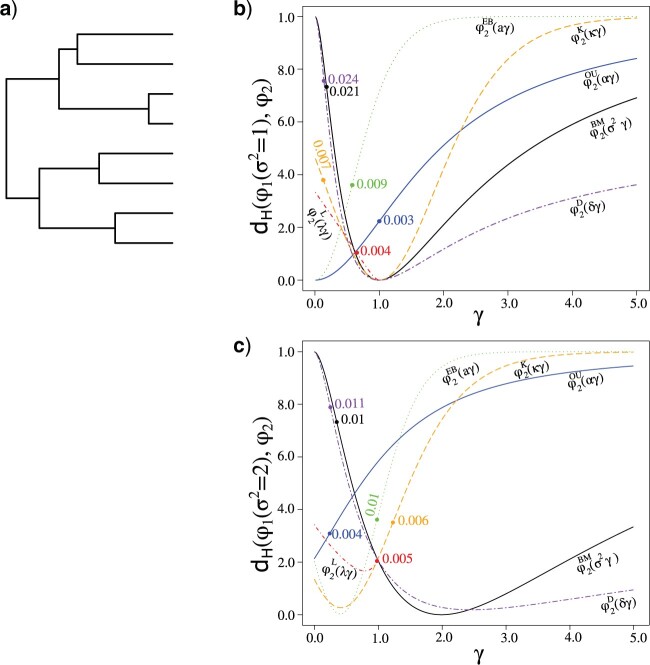
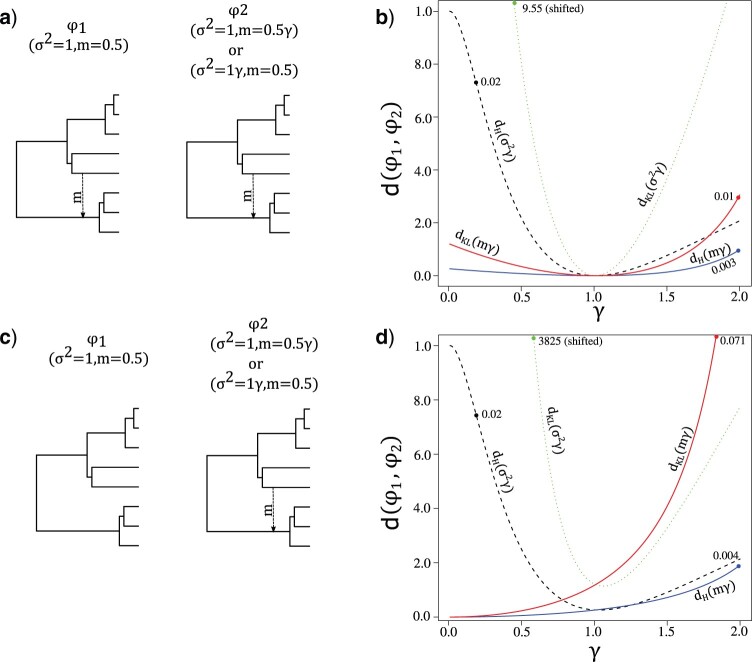
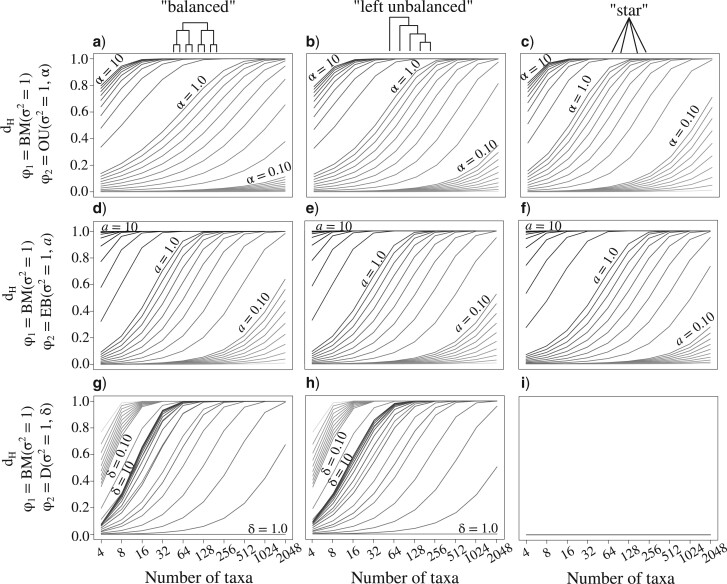
Stochastic models of character trait evolution have become a cornerstone of evolutionary biology in an array of contexts. While probabilistic models have been used extensively for statistical inference, they have largely been ignored for the purpose of measuring distances between phylogeny-aware models. Recent contributions to the problem of phylogenetic distance computation have highlighted the importance of explicitly considering evolutionary model parameters and their impacts on molecular sequence data when quantifying dissimilarity between trees. By comparing two phylogenies in terms of their induced probability distributions that are functions of many model parameters, these distances can be more informative than traditional approaches that rely strictly on differences in topology or branch lengths alone. Currently, however, these approaches are designed for comparing models of nucleotide substitution and gene tree distributions, and thus, are unable to address other classes of traits and associated models that may be of interest to evolutionary biologists. Here, we expand the principles of probabilistic phylogenetic distances to compute tree distances under models of continuous trait evolution along a phylogeny. By explicitly considering both the degree of relatedness among species and the evolutionary processes that collectively give rise to character traits, these distances provide a foundation for comparing models and their predictions, and for quantifying the impacts of assuming one phylogenetic background over another while studying the evolution of a particular trait. We demonstrate the properties of these approaches using theory, simulations, and several empirical data sets that highlight potential uses of probabilistic distances in many scenarios. We also introduce an open-source R package named PRDATR for easy application by the scientific community for computing phylogenetic distances under models of character trait evolution.[Brownian motion; comparative methods; phylogeny; quantitative traits.].
© The Author(s) 2021. Published by Oxford University Press, on behalf of the Society of Systematic Biologists.
Figures
References
-
- Abou-Moustafa K.T., Ferrie F.P.. 2012. A note on metric properties for some divergence measures: the Gaussian case. J. Mach. Learn. Res. 15:1–15.
-
- Adams R.H., Castoe T.A.. 2019a. Statistical binning leads to profound model violation due to gene tree error incurred by trying to avoid gene tree error. Mol. Phylogenet. Evol. 134:164–171. - PubMed
-
- Adams R.H., Castoe T.A.. 2019b. Probabilistic species tree distances: implementing the multispecies coalescent to compare species trees within the same model-based framework used to estimate them. Syst. Biol. 61:194–207. - PubMed
-
- Akaike H. 1973. Information theory and an extension of the maximum likelihood principle. 2nd International Symposium on Information Theory. Budapest: Akademiai Kiado. p. 267–281.
-
- Aldous D.J. 1995. Probability distributions on cladograms. In: Aldous D.J., Pemantle R., editors. Random discrete structures. Berlin: Springer. p. 1–18.
Publication types
MeSH terms
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
