

WACS: improving ChIP-seq peak calling by optimally weighting controls
- PMID: 33588754
- PMCID: PMC7885521
- DOI: 10.1186/s12859-020-03927-2
WACS: improving ChIP-seq peak calling by optimally weighting controls
Abstract
Background: Chromatin immunoprecipitation followed by high throughput sequencing (ChIP-seq), initially introduced more than a decade ago, is widely used by the scientific community to detect protein/DNA binding and histone modifications across the genome. Every experiment is prone to noise and bias, and ChIP-seq experiments are no exception. To alleviate bias, the incorporation of control datasets in ChIP-seq analysis is an essential step. The controls are used to account for the background signal, while the remainder of the ChIP-seq signal captures true binding or histone modification. However, a recurrent issue is different types of bias in different ChIP-seq experiments. Depending on which controls are used, different aspects of ChIP-seq bias are better or worse accounted for, and peak calling can produce different results for the same ChIP-seq experiment. Consequently, generating "smart" controls, which model the non-signal effect for a specific ChIP-seq experiment, could enhance contrast and increase the reliability and reproducibility of the results.
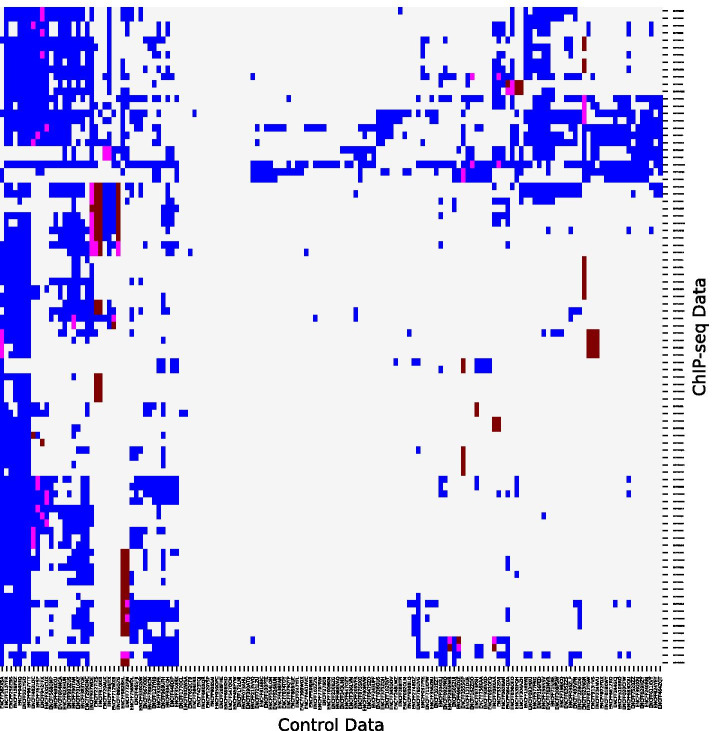
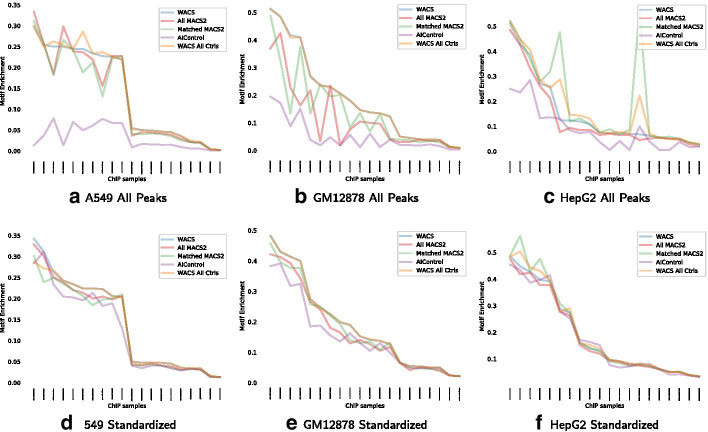
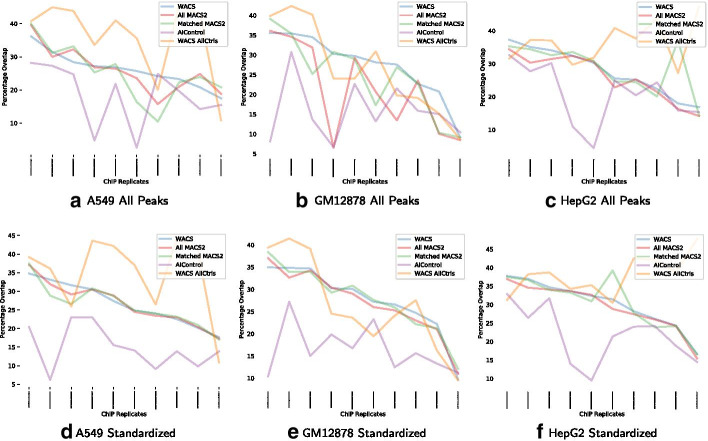
Result: We propose a peak calling algorithm, Weighted Analysis of ChIP-seq (WACS), which is an extension of the well-known peak caller MACS2. There are two main steps in WACS: First, weights are estimated for each control using non-negative least squares regression. The goal is to customize controls to model the noise distribution for each ChIP-seq experiment. This is then followed by peak calling. We demonstrate that WACS significantly outperforms MACS2 and AIControl, another recent algorithm for generating smart controls, in the detection of enriched regions along the genome, in terms of motif enrichment and reproducibility analyses.
Conclusions: This ultimately improves our understanding of ChIP-seq controls and their biases, and shows that WACS results in a better approximation of the noise distribution in controls.
Keywords: Bias; ChIP-seq; Controls.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
