

Dopamine regulates pancreatic glucagon and insulin secretion via adrenergic and dopaminergic receptors
- PMID: 33589583
- PMCID: PMC7884786
- DOI: 10.1038/s41398-020-01171-z
Dopamine regulates pancreatic glucagon and insulin secretion via adrenergic and dopaminergic receptors
Abstract
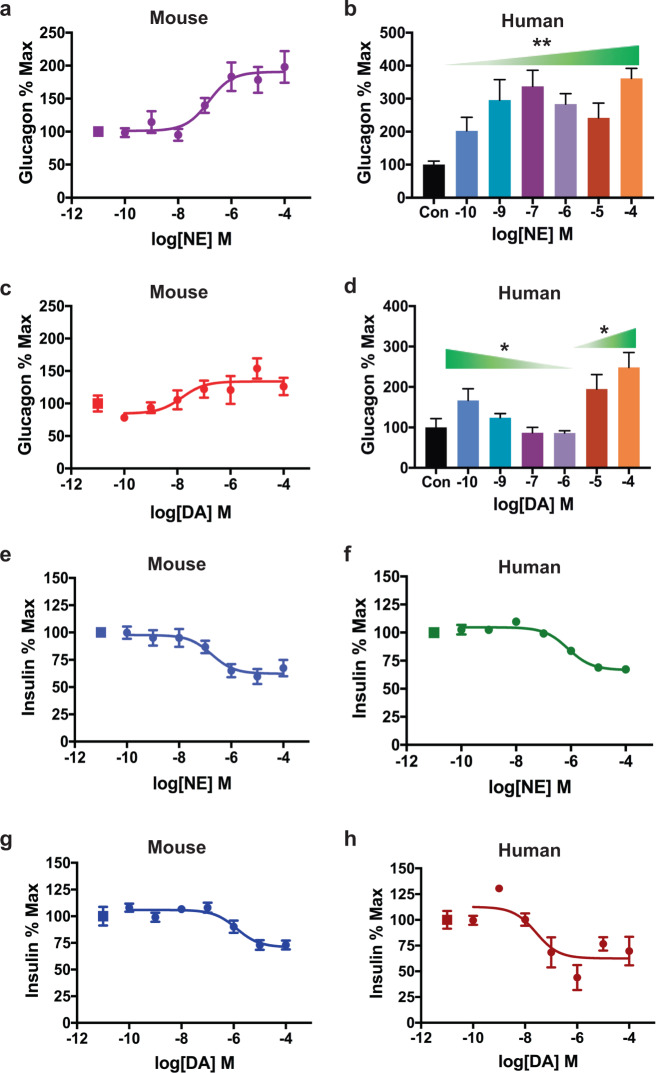
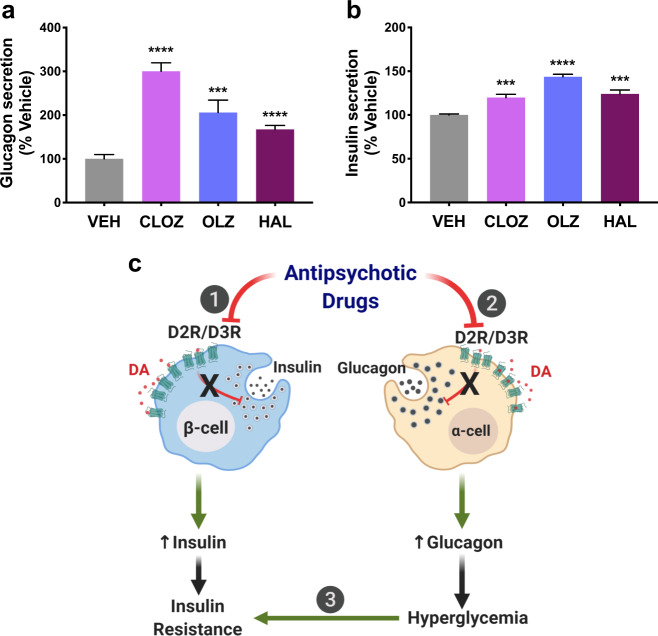
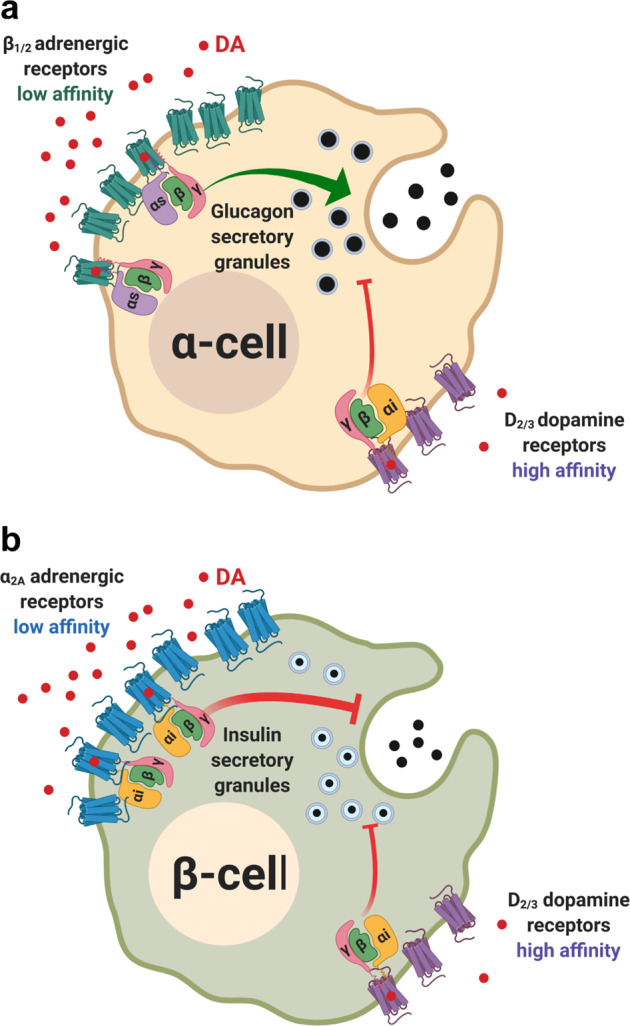
Dopamine (DA) and norepinephrine (NE) are catecholamines primarily studied in the central nervous system that also act in the pancreas as peripheral regulators of metabolism. Pancreatic catecholamine signaling has also been increasingly implicated as a mechanism responsible for the metabolic disturbances produced by antipsychotic drugs (APDs). Critically, however, the mechanisms by which catecholamines modulate pancreatic hormone release are not completely understood. We show that human and mouse pancreatic α- and β-cells express the catecholamine biosynthetic and signaling machinery, and that α-cells synthesize DA de novo. This locally-produced pancreatic DA signals via both α- and β-cell adrenergic and dopaminergic receptors with different affinities to regulate glucagon and insulin release. Significantly, we show DA functions as a biased agonist at α2A-adrenergic receptors, preferentially signaling via the canonical G protein-mediated pathway. Our findings highlight the interplay between DA and NE signaling as a novel form of regulation to modulate pancreatic hormone release. Lastly, pharmacological blockade of DA D2-like receptors in human islets with APDs significantly raises insulin and glucagon release. This offers a new mechanism where APDs act directly on islet α- and β-cell targets to produce metabolic disturbances.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures


References
Publication types
MeSH terms
Substances
Grants and funding
- R01 DK097160/DK/NIDDK NIH HHS/United States
- PR141292/U.S. Department of Defense (United States Department of Defense)
- R21 AG068607/AG/NIA NIH HHS/United States
- K08 DA031241/DA/NIDA NIH HHS/United States
- R01DK097160/U.S. Department of Health & Human Services | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (National Institute of Diabetes & Digestive & Kidney Diseases)
LinkOut - more resources
Full Text Sources
Other Literature Sources
