

Causal network models of SARS-CoV-2 expression and aging to identify candidates for drug repurposing
- PMID: 33589624
- PMCID: PMC7884845
- DOI: 10.1038/s41467-021-21056-z
Causal network models of SARS-CoV-2 expression and aging to identify candidates for drug repurposing
Abstract
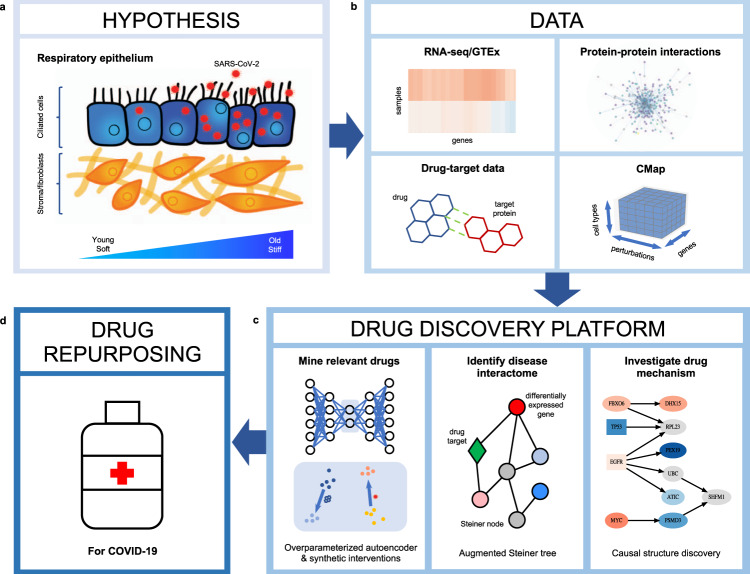
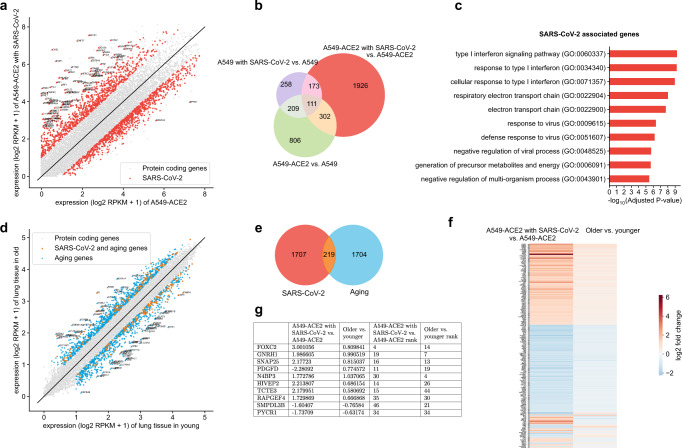
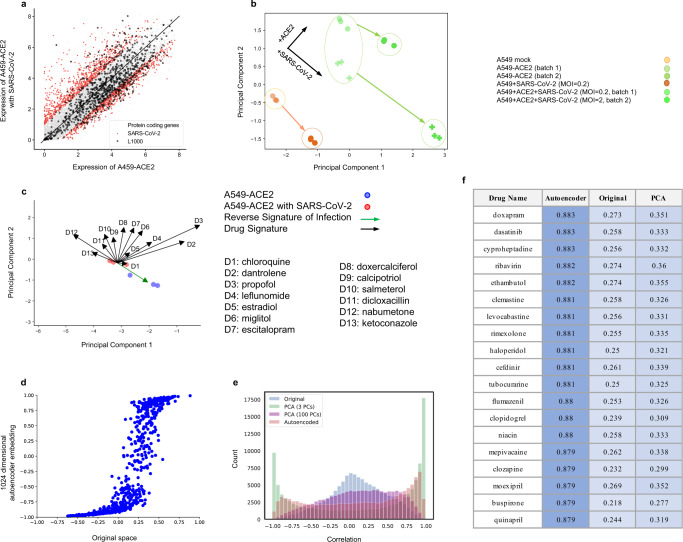
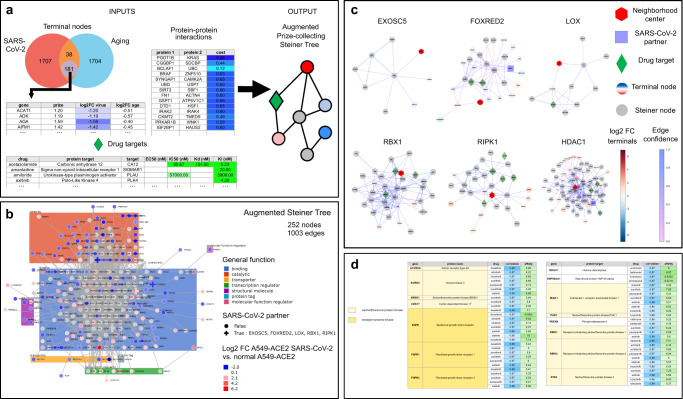
Given the severity of the SARS-CoV-2 pandemic, a major challenge is to rapidly repurpose existing approved drugs for clinical interventions. While a number of data-driven and experimental approaches have been suggested in the context of drug repurposing, a platform that systematically integrates available transcriptomic, proteomic and structural data is missing. More importantly, given that SARS-CoV-2 pathogenicity is highly age-dependent, it is critical to integrate aging signatures into drug discovery platforms. We here take advantage of large-scale transcriptional drug screens combined with RNA-seq data of the lung epithelium with SARS-CoV-2 infection as well as the aging lung. To identify robust druggable protein targets, we propose a principled causal framework that makes use of multiple data modalities. Our analysis highlights the importance of serine/threonine and tyrosine kinases as potential targets that intersect the SARS-CoV-2 and aging pathways. By integrating transcriptomic, proteomic and structural data that is available for many diseases, our drug discovery platform is broadly applicable. Rigorous in vitro experiments as well as clinical trials are needed to validate the identified candidate drugs.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Lung disease network reveals impact of comorbidity on SARS-CoV-2 infection and opportunities of drug repurposing.BMC Med Genomics. 2021 Sep 17;14(1):226. doi: 10.1186/s12920-021-01079-7. BMC Med Genomics. 2021. PMID: 34535131 Free PMC article.
-
Mapping the SARS-CoV-2-Host Protein-Protein Interactome by Affinity Purification Mass Spectrometry and Proximity-Dependent Biotin Labeling: A Rational and Straightforward Route to Discover Host-Directed Anti-SARS-CoV-2 Therapeutics.Int J Mol Sci. 2021 Jan 7;22(2):532. doi: 10.3390/ijms22020532. Int J Mol Sci. 2021. PMID: 33430309 Free PMC article. Review.
-
Comparative transcriptome analysis of SARS-CoV, MERS-CoV, and SARS-CoV-2 to identify potential pathways for drug repurposing.Comput Biol Med. 2021 Jan;128:104123. doi: 10.1016/j.compbiomed.2020.104123. Epub 2020 Nov 24. Comput Biol Med. 2021. PMID: 33260034 Free PMC article.
-
Predicting Potential SARS-COV-2 Drugs-In Depth Drug Database Screening Using Deep Neural Network Framework SSnet, Classical Virtual Screening and Docking.Int J Mol Sci. 2021 Feb 4;22(4):1573. doi: 10.3390/ijms22041573. Int J Mol Sci. 2021. PMID: 33557253 Free PMC article.
-
Role of ACE2 receptor and the landscape of treatment options from convalescent plasma therapy to the drug repurposing in COVID-19.Mol Cell Biochem. 2021 Feb;476(2):553-574. doi: 10.1007/s11010-020-03924-2. Epub 2020 Oct 7. Mol Cell Biochem. 2021. PMID: 33029696 Free PMC article. Review.
Cited by
-
Causal reasoning over knowledge graphs leveraging drug-perturbed and disease-specific transcriptomic signatures for drug discovery.PLoS Comput Biol. 2022 Feb 25;18(2):e1009909. doi: 10.1371/journal.pcbi.1009909. eCollection 2022 Feb. PLoS Comput Biol. 2022. PMID: 35213534 Free PMC article.
-
Image2Reg: Linking chromatin images to gene regulation using genetic and chemical perturbation screens.Cell Syst. 2025 Jun 18;16(6):101293. doi: 10.1016/j.cels.2025.101293. Epub 2025 May 12. Cell Syst. 2025. PMID: 40359941
-
Local patterns of genetic sharing challenge the boundaries between neuropsychiatric and insulin resistance-related conditions.medRxiv [Preprint]. 2024 Mar 8:2024.03.07.24303921. doi: 10.1101/2024.03.07.24303921. medRxiv. 2024. Update in: Transl Psychiatry. 2025 Apr 12;15(1):145. doi: 10.1038/s41398-025-03349-9. PMID: 38496672 Free PMC article. Updated. Preprint.
-
A multilevel approach for screening natural compounds as an antiviral agent for COVID-19.Comput Biol Chem. 2022 Jun;98:107694. doi: 10.1016/j.compbiolchem.2022.107694. Epub 2022 May 11. Comput Biol Chem. 2022. PMID: 35576744 Free PMC article.
-
Strategies to identify candidate repurposable drugs: COVID-19 treatment as a case example.Transl Psychiatry. 2021 Nov 16;11(1):591. doi: 10.1038/s41398-021-01724-w. Transl Psychiatry. 2021. PMID: 34785660 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
