

Fast and precise single-cell data analysis using a hierarchical autoencoder
- PMID: 33589635
- PMCID: PMC7884436
- DOI: 10.1038/s41467-021-21312-2
Fast and precise single-cell data analysis using a hierarchical autoencoder
Abstract
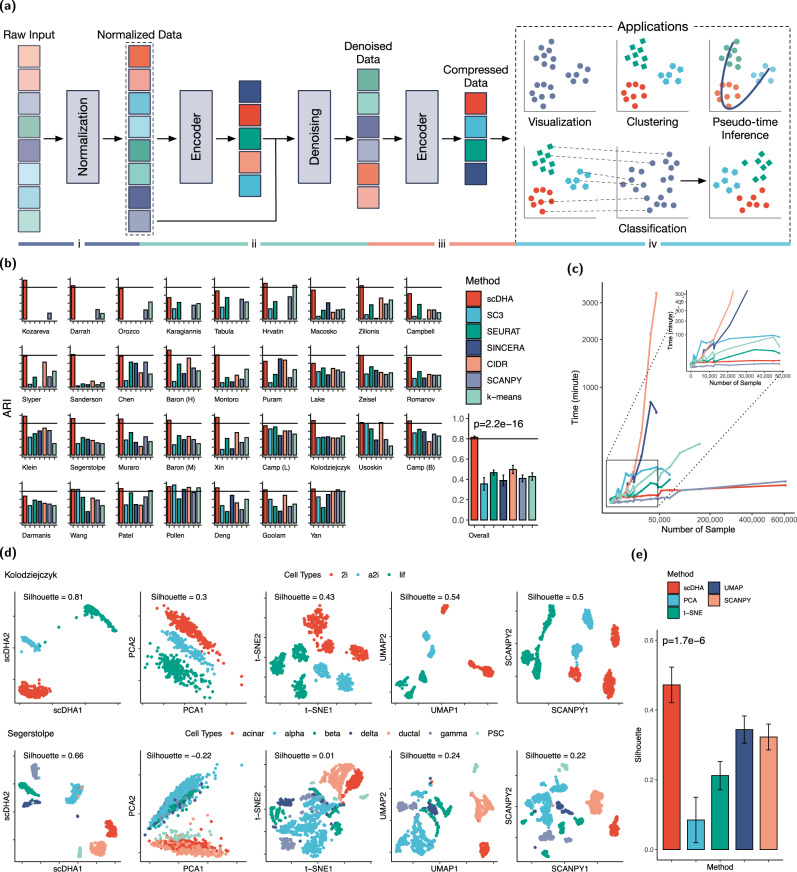
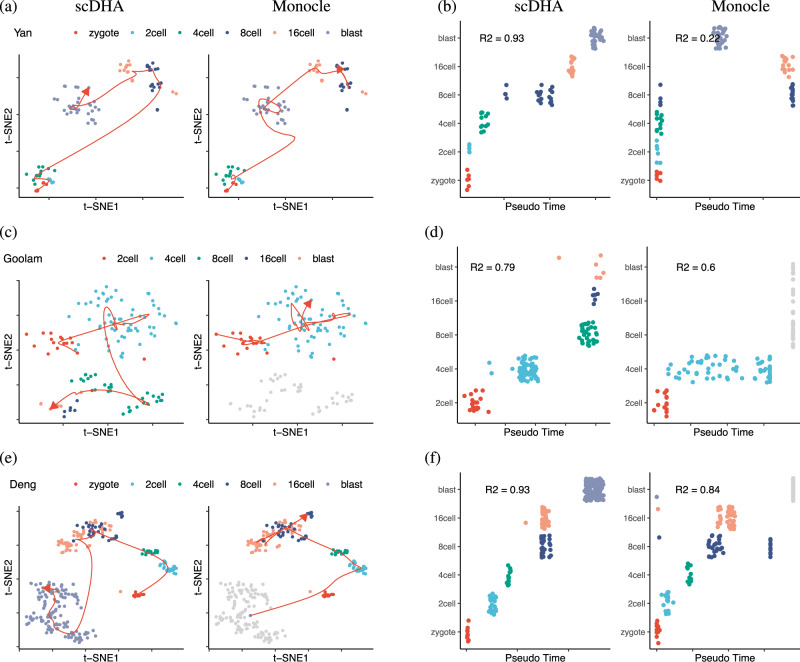
A primary challenge in single-cell RNA sequencing (scRNA-seq) studies comes from the massive amount of data and the excess noise level. To address this challenge, we introduce an analysis framework, named single-cell Decomposition using Hierarchical Autoencoder (scDHA), that reliably extracts representative information of each cell. The scDHA pipeline consists of two core modules. The first module is a non-negative kernel autoencoder able to remove genes or components that have insignificant contributions to the part-based representation of the data. The second module is a stacked Bayesian autoencoder that projects the data onto a low-dimensional space (compressed). To diminish the tendency to overfit of neural networks, we repeatedly perturb the compressed space to learn a more generalized representation of the data. In an extensive analysis, we demonstrate that scDHA outperforms state-of-the-art techniques in many research sub-fields of scRNA-seq analysis, including cell segregation through unsupervised learning, visualization of transcriptome landscape, cell classification, and pseudo-time inference.
Conflict of interest statement
The authors declare no competing interests.
Figures



References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
