

Balance between promiscuity and specificity in phage λ host range
- PMID: 33589767
- PMCID: PMC8319322
- DOI: 10.1038/s41396-021-00912-2
Balance between promiscuity and specificity in phage λ host range
Abstract
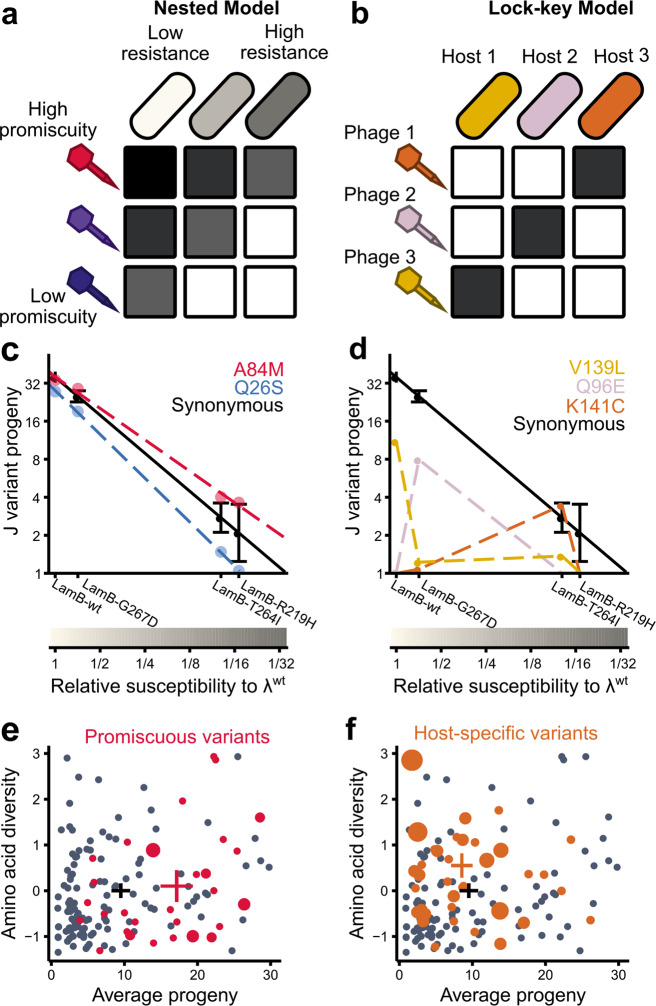
As hosts acquire resistance to viruses, viruses must overcome that resistance to re-establish infectivity, or go extinct. Despite the significant hurdles associated with adapting to a resistant host, viruses are evolutionarily successful and maintain stable coevolutionary relationships with their hosts. To investigate the factors underlying how pathogens adapt to their hosts, we performed a deep mutational scan of the region of the λ tail fiber tip protein that mediates contact with the receptor on λ's host, Escherichia coli. Phages harboring amino acid substitutions were subjected to selection for infectivity on wild type E. coli, revealing a highly restrictive fitness landscape, in which most substitutions completely abrogate function. A subset of positions that are tolerant of mutation in this assay, but diverse over evolutionary time, are associated with host range expansion. Imposing selection for phage infectivity on three λ-resistant hosts, each harboring a different missense mutation in the λ receptor, reveals hundreds of adaptive variants in λ. We distinguish λ variants that confer promiscuity, a general ability to overcome host resistance, from those that drive host-specific infectivity. Both processes may be important in driving adaptation to a novel host.
© 2021. The Author(s), under exclusive licence to International Society for Microbial Ecology.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
-
- Lenski RE, Levin BR. Constraints on the coevolution of bacteria and virulent phage: a model, some experiments, and predictions for natural communities. Am Nat. 1985;125:585–602. doi: 10.1086/284364. - DOI
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
