

Joint profiling of histone modifications and transcriptome in single cells from mouse brain
- PMID: 33589836
- PMCID: PMC7954905
- DOI: 10.1038/s41592-021-01060-3
Joint profiling of histone modifications and transcriptome in single cells from mouse brain
Abstract
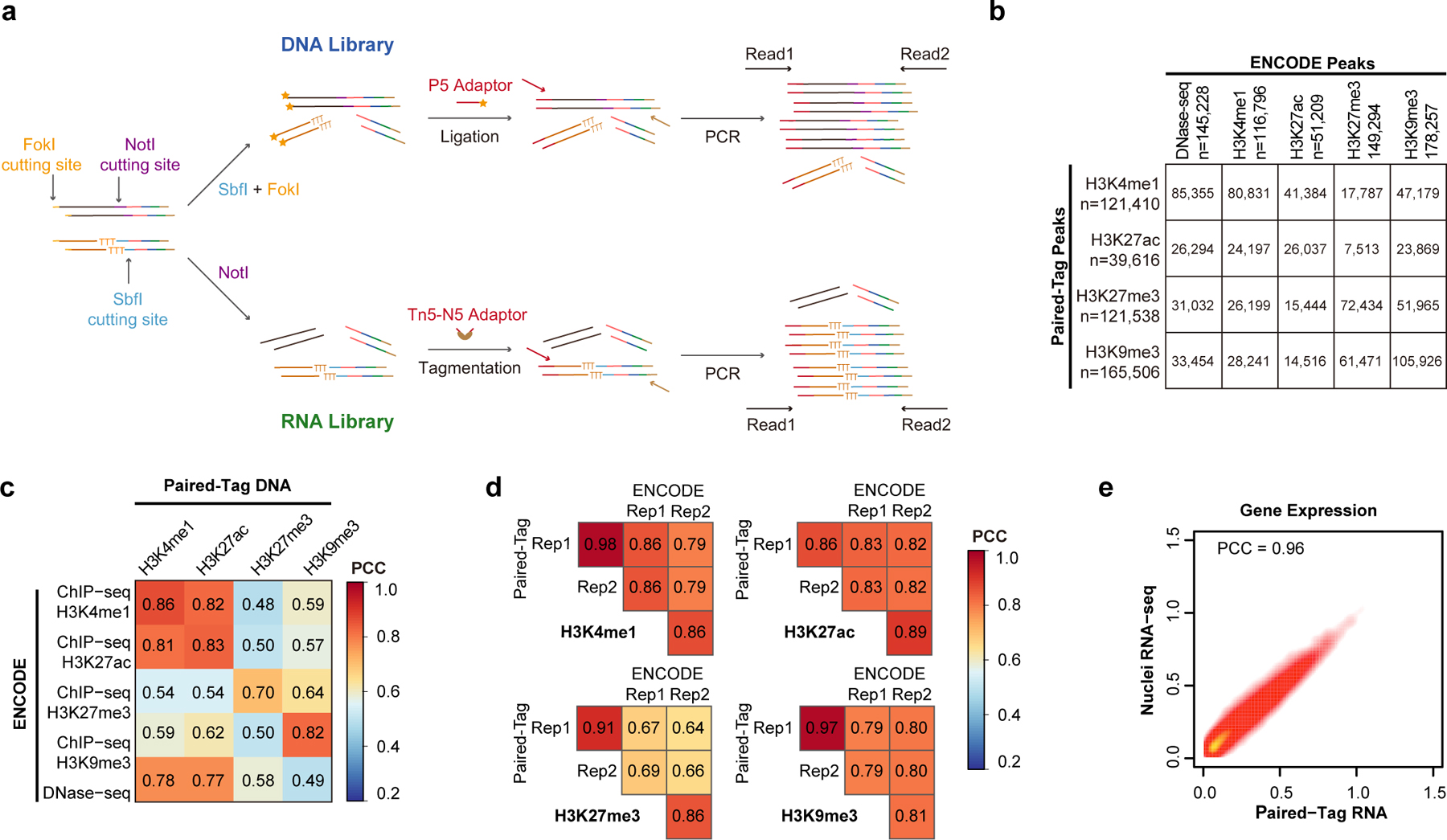
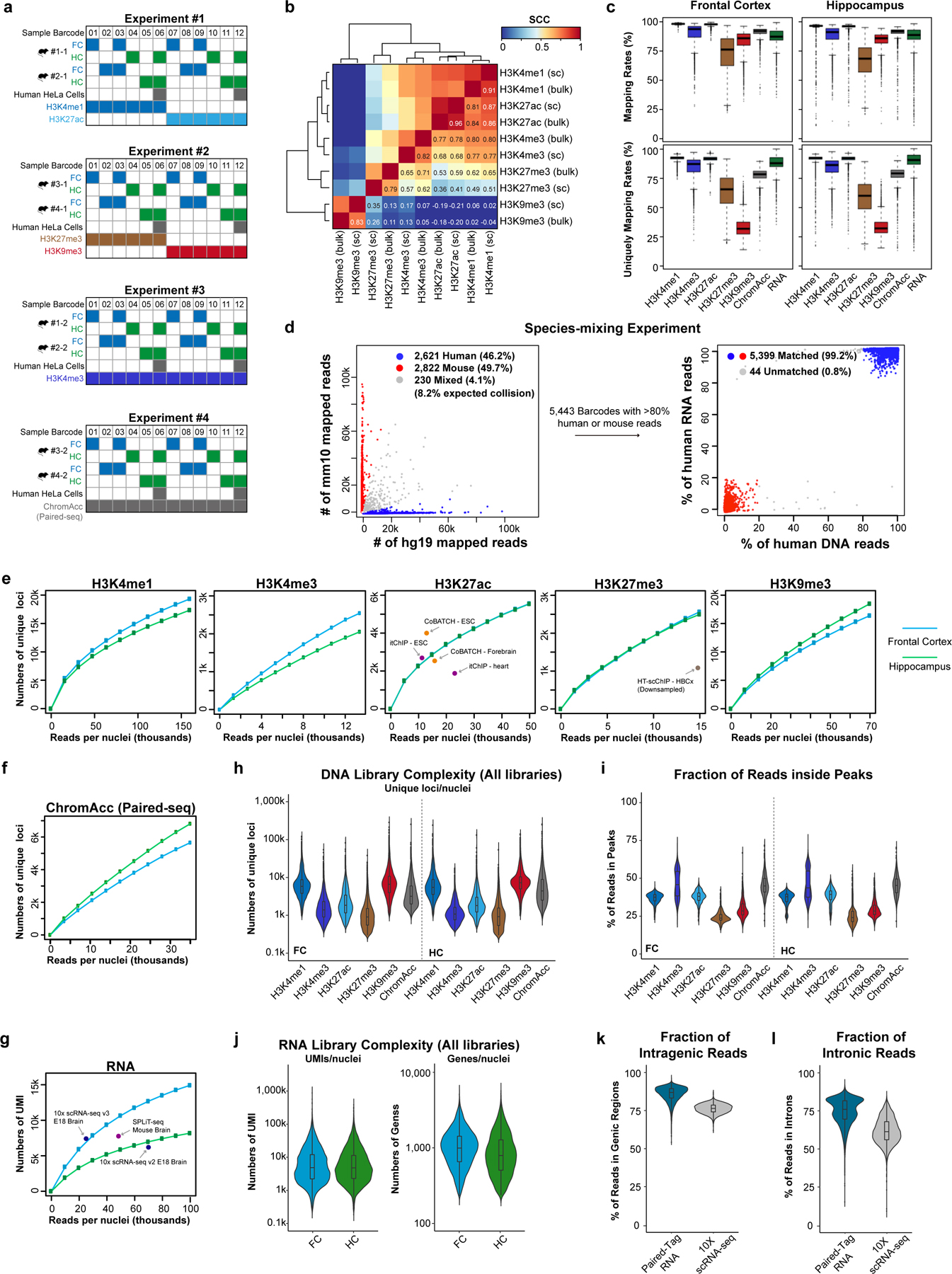
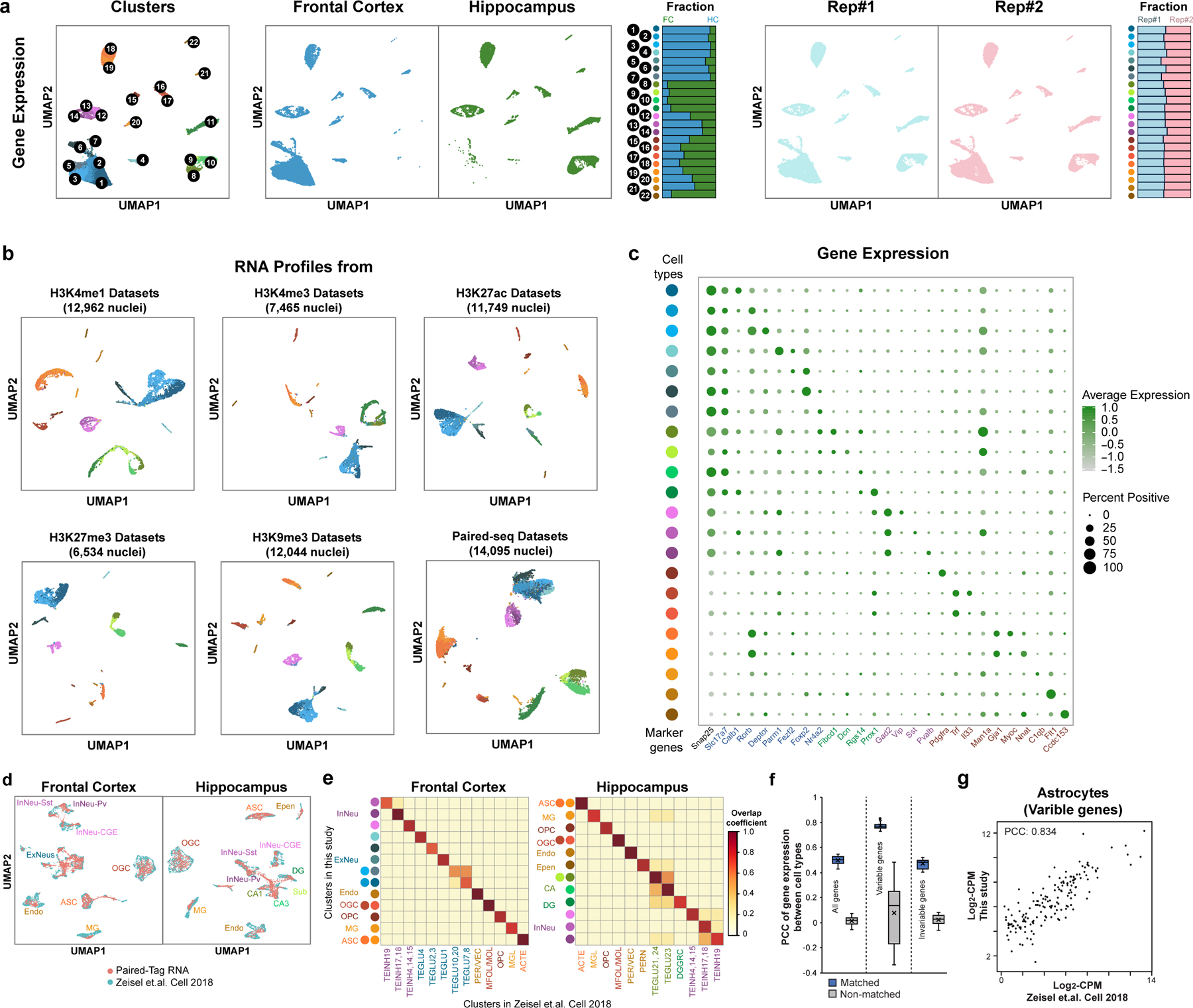
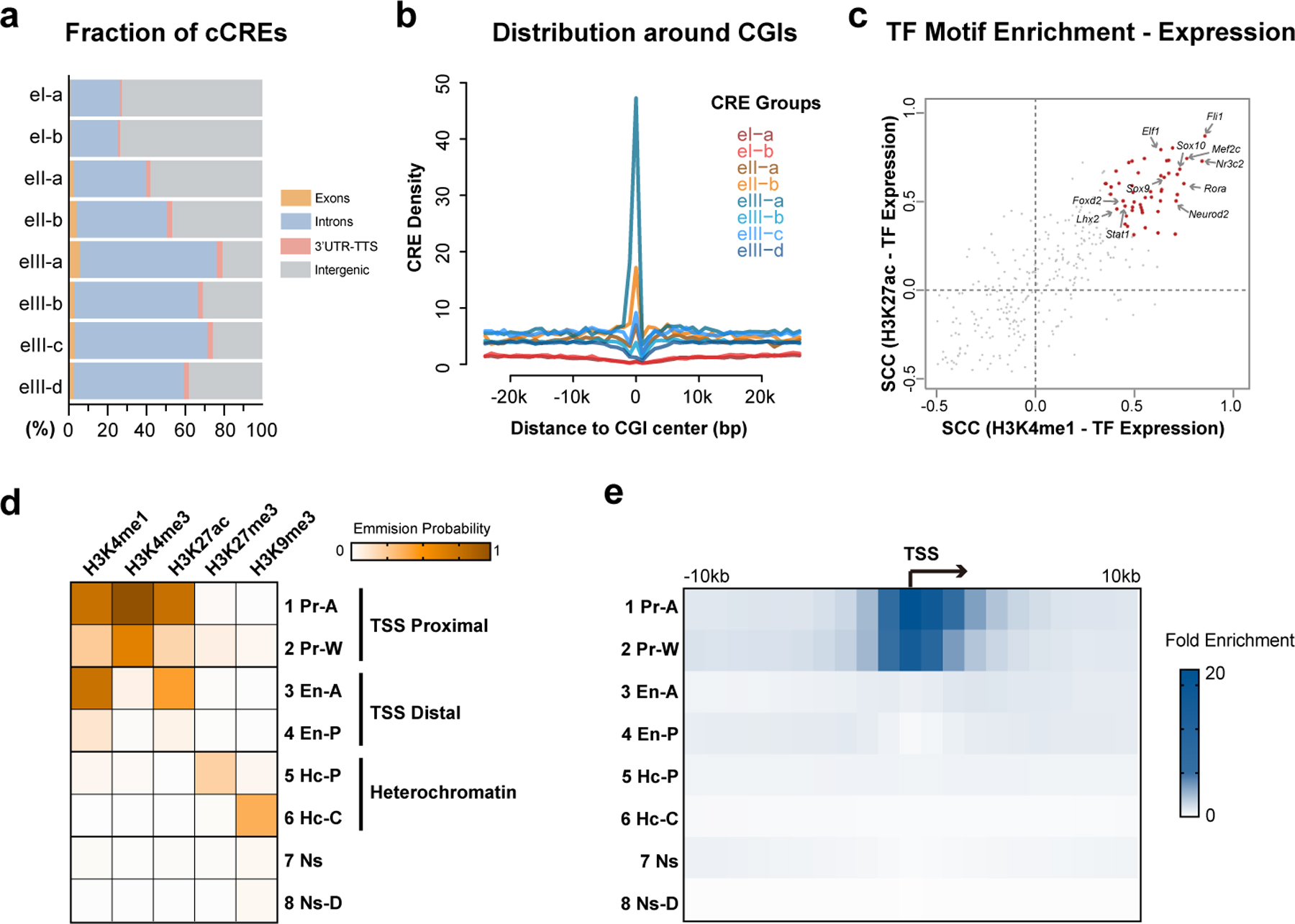
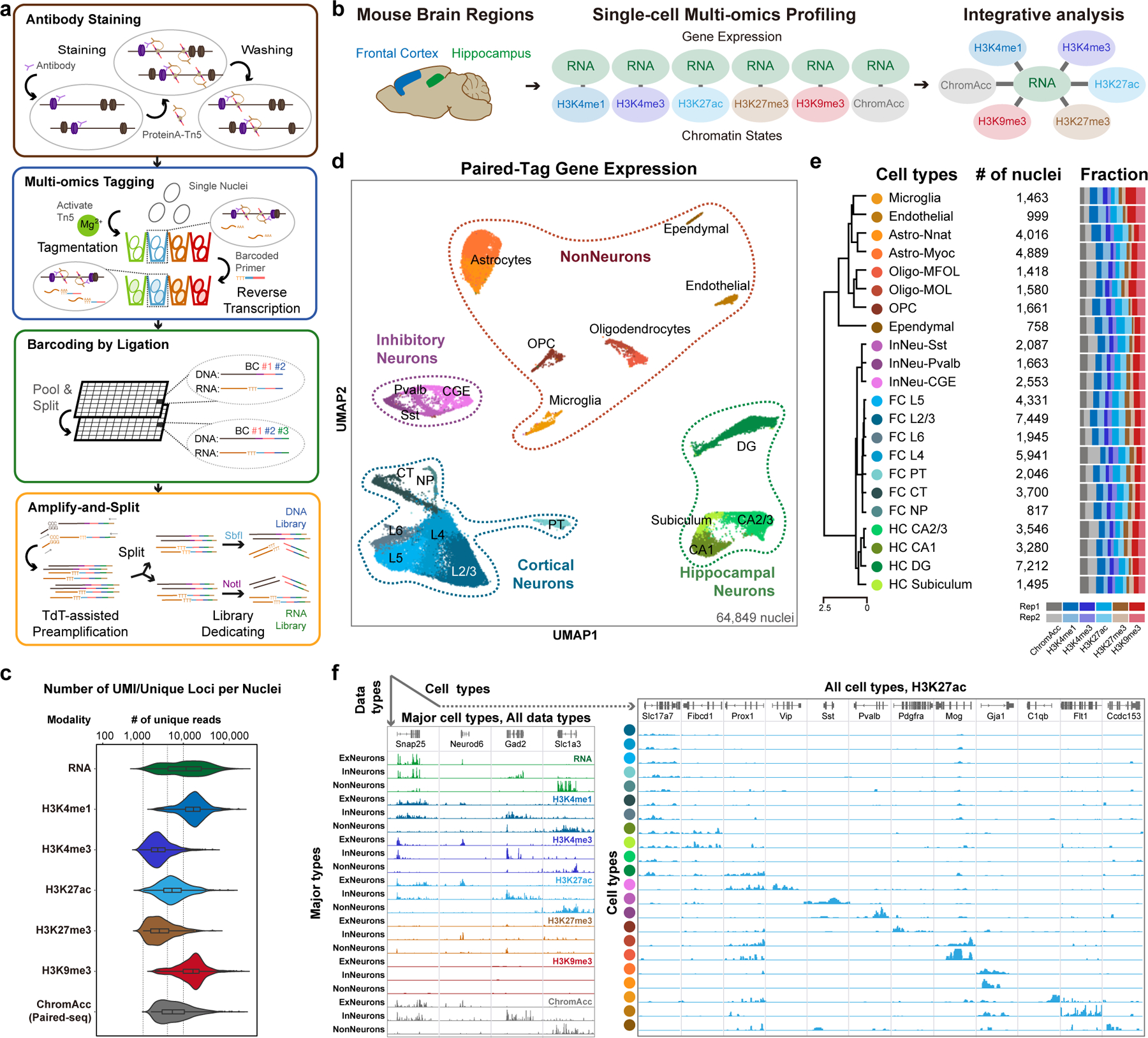
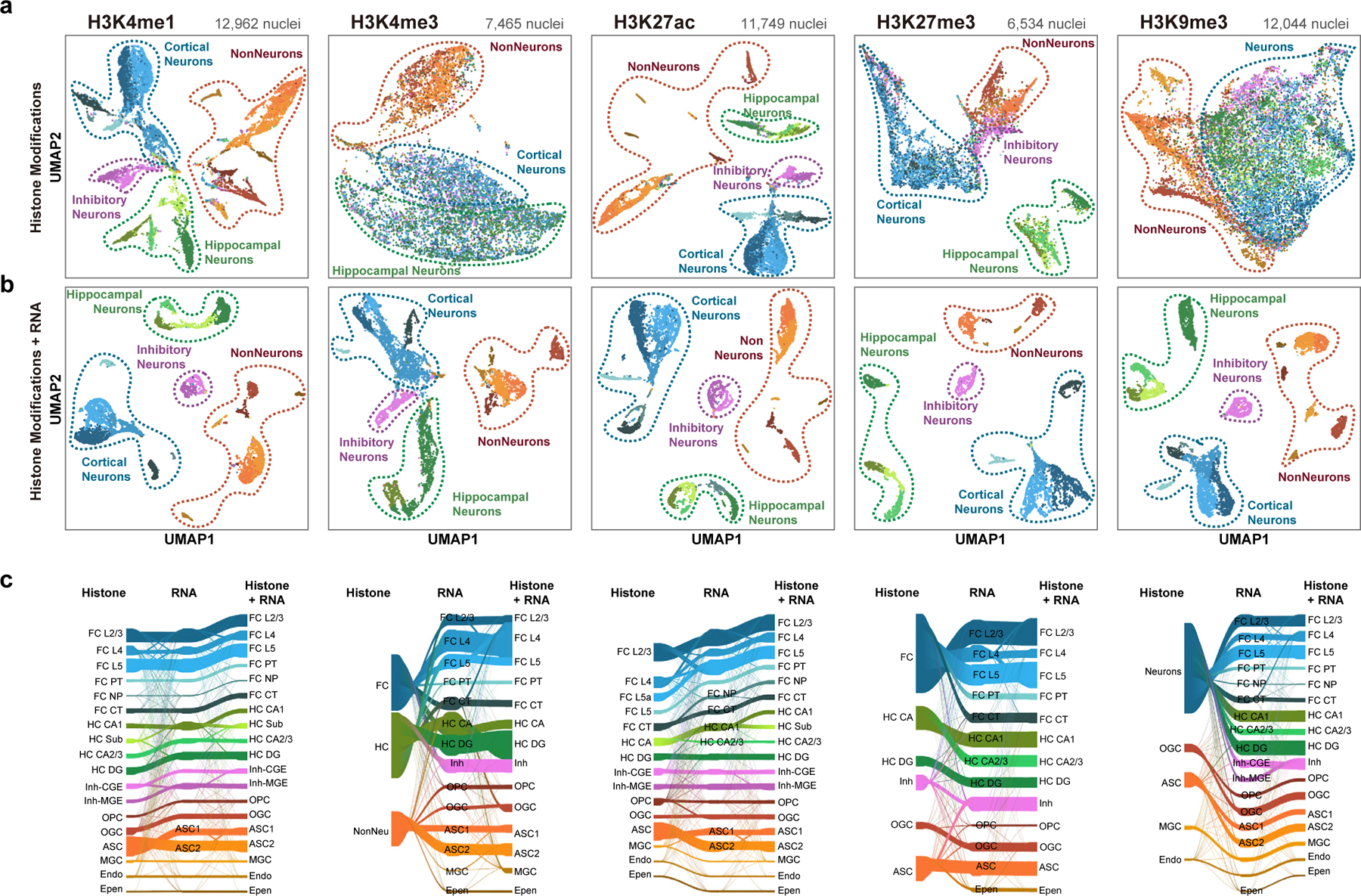
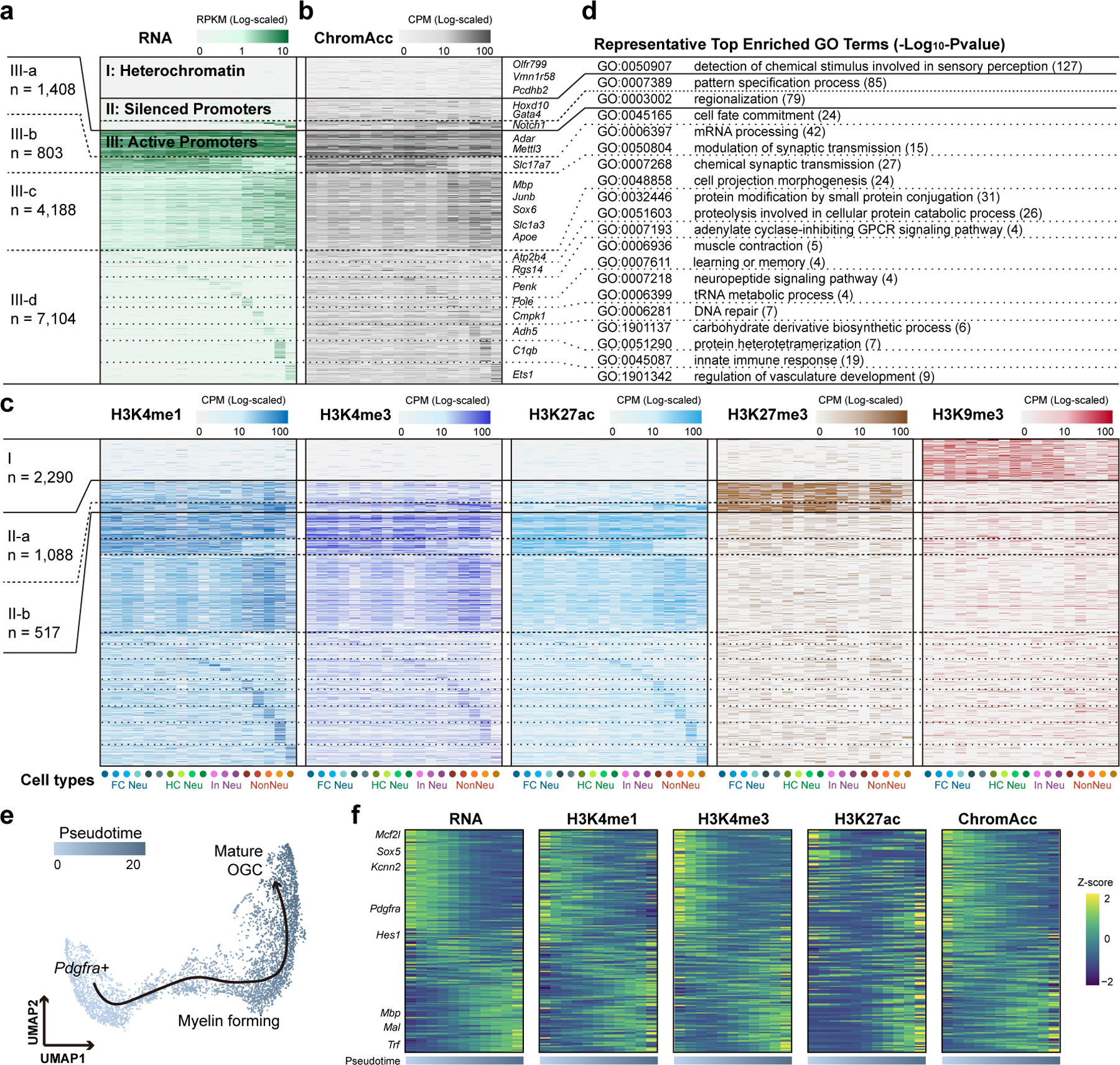
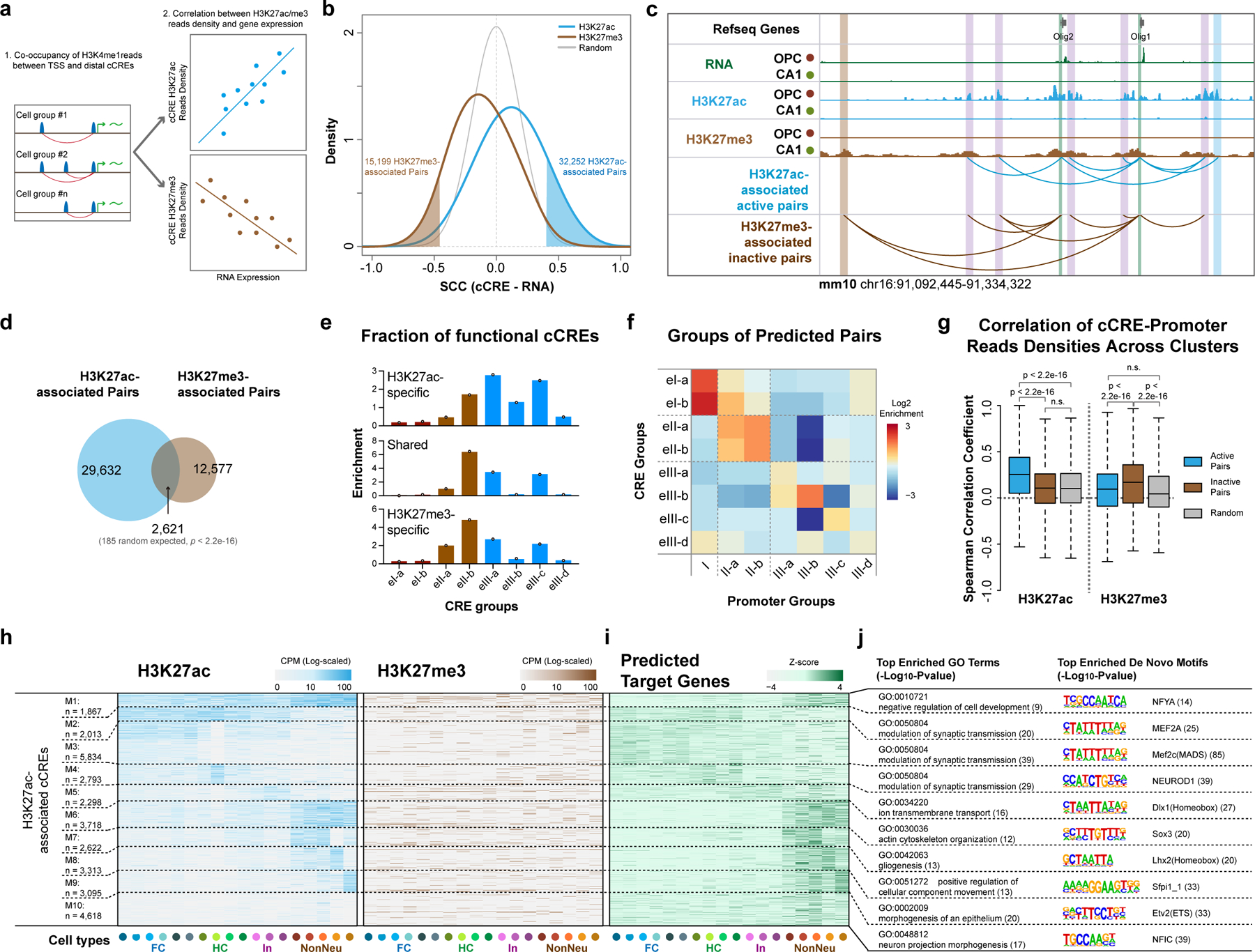
Genome-wide profiling of histone modifications can reveal not only the location and activity state of regulatory elements, but also the regulatory mechanisms involved in cell-type-specific gene expression during development and disease pathology. Conventional assays to profile histone modifications in bulk tissues lack single-cell resolution. Here we describe an ultra-high-throughput method, Paired-Tag, for joint profiling of histone modifications and transcriptome in single cells to produce cell-type-resolved maps of chromatin state and transcriptome in complex tissues. We used this method to profile five histone modifications jointly with transcriptome in the adult mouse frontal cortex and hippocampus. Integrative analysis of the resulting maps identified distinct groups of genes subject to divergent epigenetic regulatory mechanisms. Our single-cell multiomics approach enables comprehensive analysis of chromatin state and gene regulation in complex tissues and characterization of gene regulatory programs in the constituent cell types.
Conflict of interest statement
Competing interests
B.R. is a co-founder and consultant for Arima Genomics, Inc, and co-founder of Epigenome Technologies, Inc. B.R. and C.Z. are listed as inventors of a provisional patent titled “PARALLEL ANALYSIS OF INDIVIDUAL CELLS FOR RNA EXPRESSION AND DNA FROM TARGETED TAGMENTATION BY SEQUENCING”.
Figures




Comment in
-
Single-cell multiomics to probe relationships between histone modifications and transcription.Nat Methods. 2021 Jun;18(6):602-603. doi: 10.1038/s41592-021-01147-x. Nat Methods. 2021. PMID: 33958789 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
