

sangeranalyseR: Simple and Interactive Processing of Sanger Sequencing Data in R
- PMID: 33591316
- PMCID: PMC7939931
- DOI: 10.1093/gbe/evab028
sangeranalyseR: Simple and Interactive Processing of Sanger Sequencing Data in R
Abstract
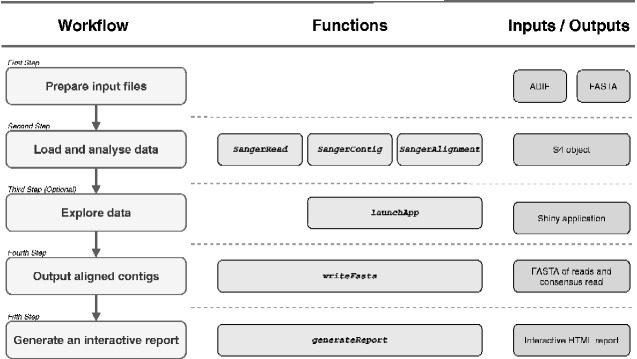
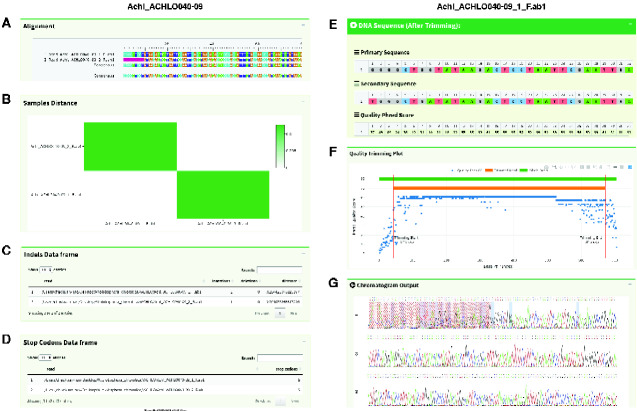
sangeranalyseR is feature-rich, free, and open-source R package for processing Sanger sequencing data. It allows users to go from loading reads to saving aligned contigs in a few lines of R code by using sensible defaults for most actions. It also provides complete flexibility for determining how individual reads and contigs are processed, both at the command-line in R and via interactive Shiny applications. sangeranalyseR provides a wide range of options for all steps in Sanger processing pipelines including trimming reads, detecting secondary peaks, viewing chromatograms, detecting indels and stop codons, aligning contigs, estimating phylogenetic trees, and more. Input data can be in either ABIF or FASTA format. sangeranalyseR comes with extensive online documentation and outputs aligned and unaligned reads and contigs in FASTA format, along with detailed interactive HTML reports. sangeranalyseR supports the use of colorblind-friendly palettes for viewing alignments and chromatograms. It is released under an MIT licence and available for all platforms on Bioconductor (https://bioconductor.org/packages/sangeranalyseR, last accessed February 22, 2021) and on Github (https://github.com/roblanf/sangeranalyseR, last accessed February 22, 2021).
Keywords: DNA; alignment; bioconductor; chromatogram; genetics; shiny application.
© The Author(s) 2021. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures

References
-
- Allaire J, Xie Y, McPherson J, Luraschi J, Ushey K, Atkins A, Wickham H, Cheng J, Chang W, Iannone R (2021). rmarkdown: Dynamic Documents for R. R package version 2.7, https://github.com/rstudio/rmarkdown
-
- Attali D. 2020. shinyjs: easily improve the user experience of your shiny apps in seconds. R package version 2.0.0. Available from: https://cran.r-project.org/web/packages/shinyjs/index.html
-
- Ewing B, Green P.. 1998. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 8(3):186–194. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
