

Endoplasmic reticulum-associated degradation is required for nephrin maturation and kidney glomerular filtration function
- PMID: 33591954
- PMCID: PMC8011890
- DOI: 10.1172/JCI143988
Endoplasmic reticulum-associated degradation is required for nephrin maturation and kidney glomerular filtration function
Abstract
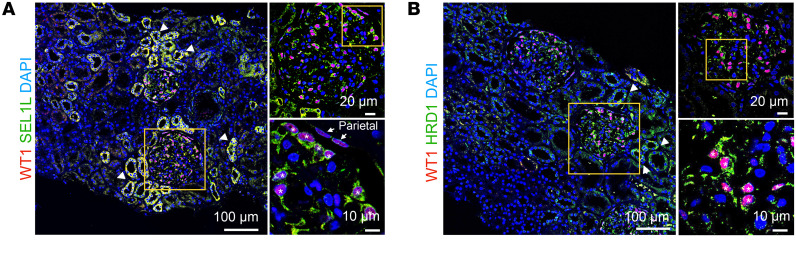
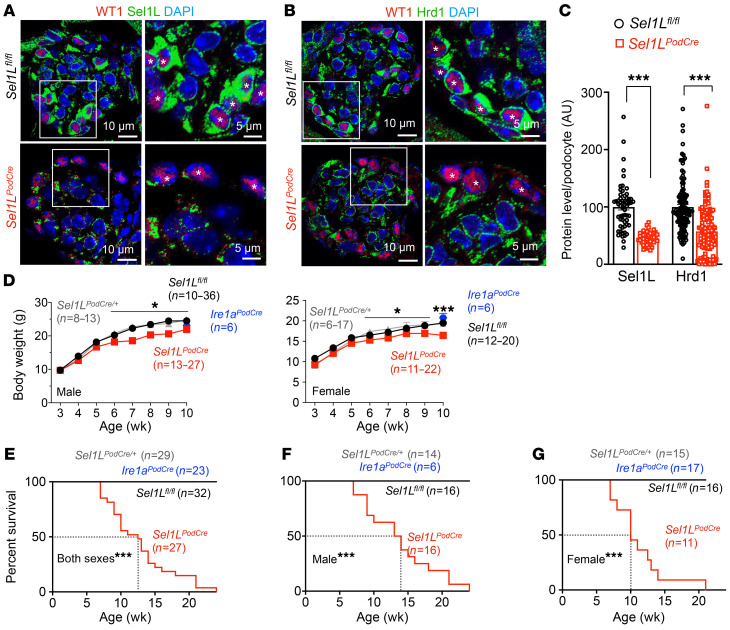
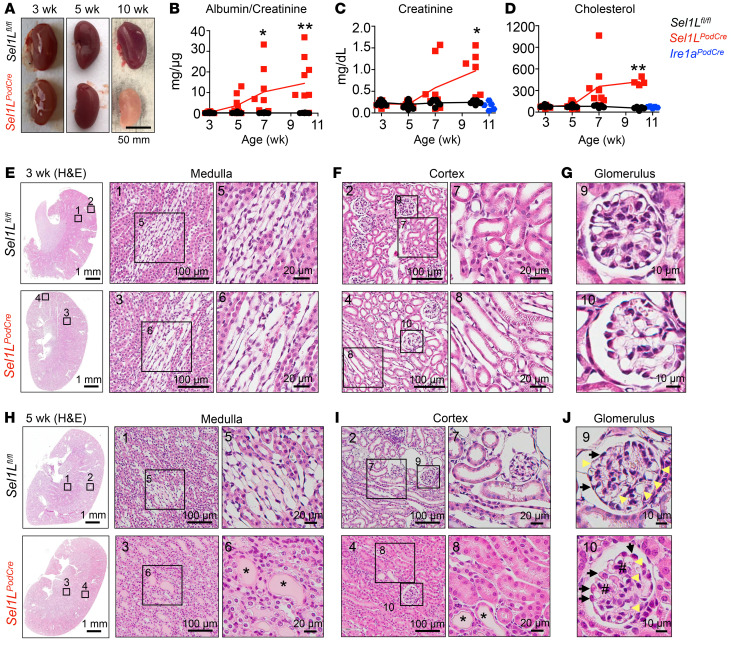
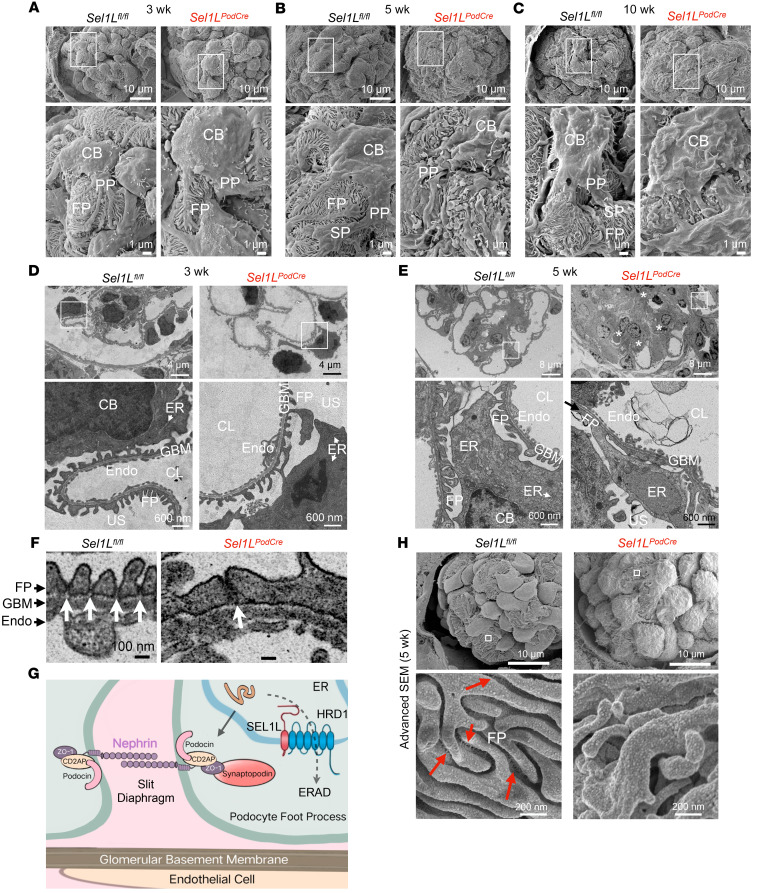
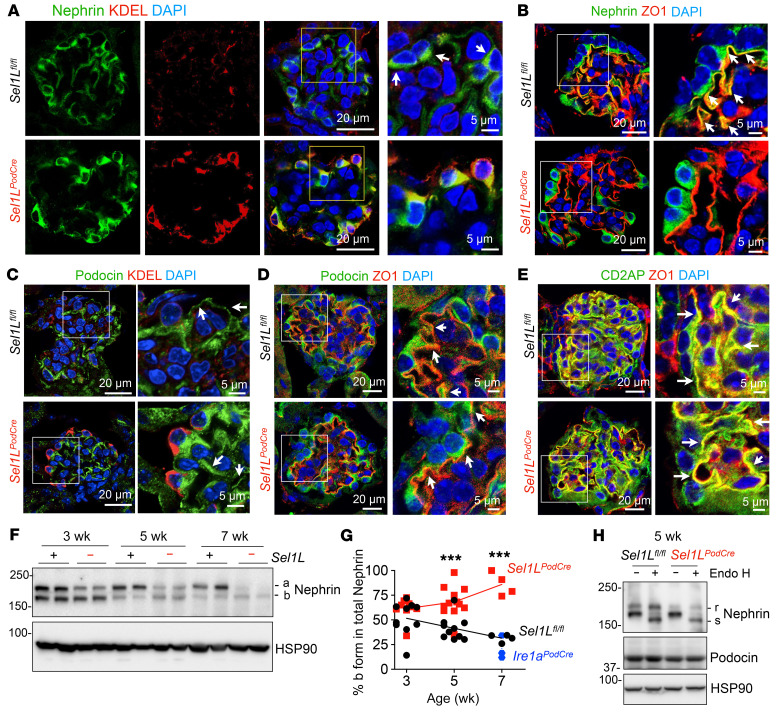
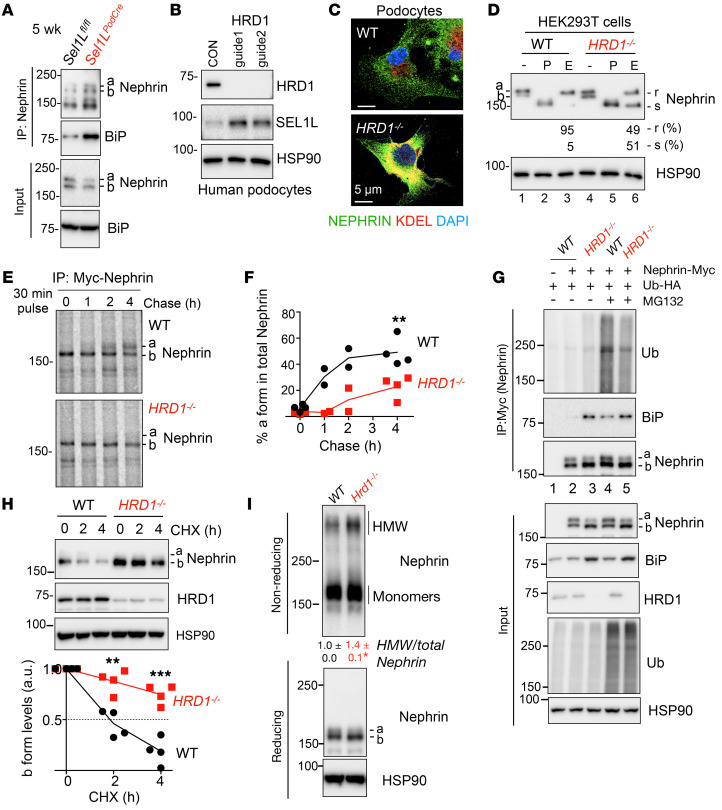
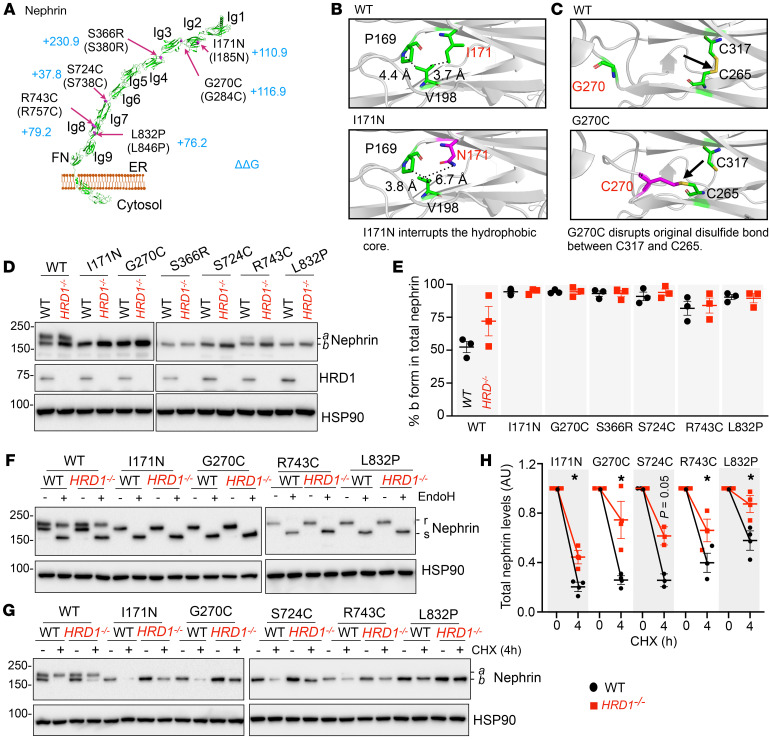
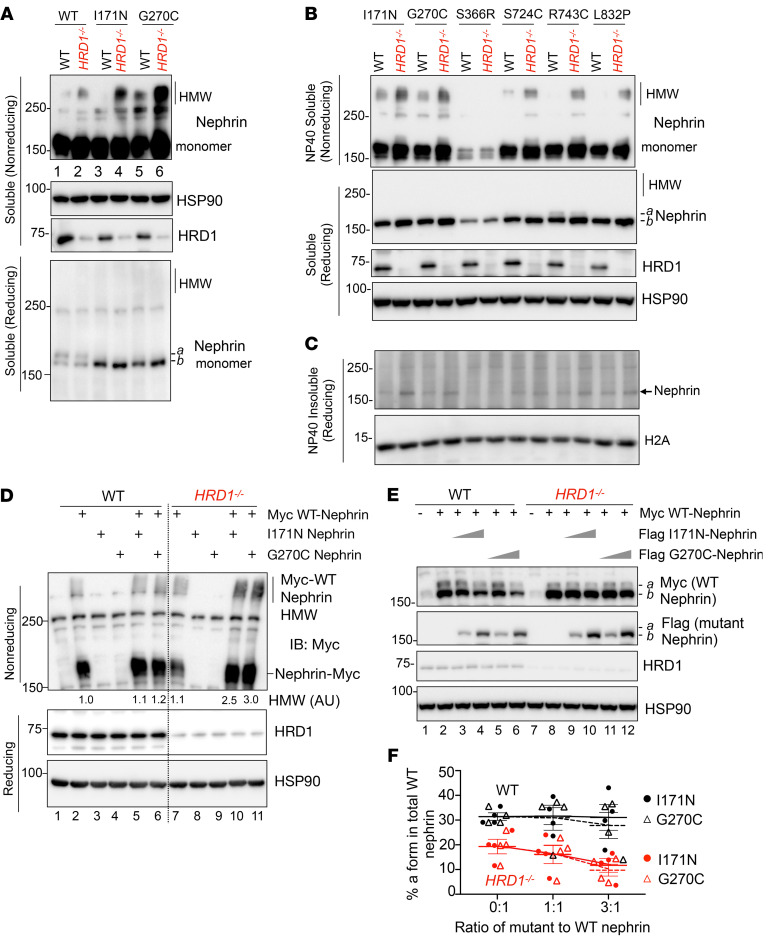
Podocytes are key to the glomerular filtration barrier by forming a slit diaphragm between interdigitating foot processes; however, the molecular details and functional importance of protein folding and degradation in the ER remain unknown. Here, we show that the SEL1L-HRD1 protein complex of ER-associated degradation (ERAD) is required for slit diaphragm formation and glomerular filtration function. SEL1L-HRD1 ERAD is highly expressed in podocytes of both mouse and human kidneys. Mice with podocyte-specific Sel1L deficiency develop podocytopathy and severe congenital nephrotic syndrome with an impaired slit diaphragm shortly after weaning and die prematurely, with a median lifespan of approximately 3 months. We show mechanistically that nephrin, a type 1 membrane protein causally linked to congenital nephrotic syndrome, is an endogenous ERAD substrate. ERAD deficiency attenuated the maturation of nascent nephrin, leading to its retention in the ER. We also show that various autosomal-recessive nephrin disease mutants were highly unstable and broken down by SEL1L-HRD1 ERAD, which attenuated the pathogenicity of the mutants toward the WT allele. This study uncovers a critical role of SEL1L-HRD1 ERAD in glomerular filtration barrier function and provides insights into the pathogenesis associated with autosomal-recessive disease mutants.
Keywords: Cell Biology; Nephrology; Protein misfolding; Protein traffic; Ubiquitin-proteosome system.
Conflict of interest statement
Figures
Similar articles
-
Regulation of hepatic inclusions and fibrinogen biogenesis by SEL1L-HRD1 ERAD.Nat Commun. 2024 Oct 26;15(1):9244. doi: 10.1038/s41467-024-53639-x. Nat Commun. 2024. PMID: 39455574 Free PMC article.
-
Hepatic SEL1L-HRD1 ER-associated degradation regulates systemic iron homeostasis via ceruloplasmin.Proc Natl Acad Sci U S A. 2023 Jan 10;120(2):e2212644120. doi: 10.1073/pnas.2212644120. Epub 2023 Jan 3. Proc Natl Acad Sci U S A. 2023. PMID: 36595688 Free PMC article.
-
Hepatic Sel1L-Hrd1 ER-associated degradation (ERAD) manages FGF21 levels and systemic metabolism via CREBH.EMBO J. 2018 Nov 15;37(22):e99277. doi: 10.15252/embj.201899277. Epub 2018 Nov 2. EMBO J. 2018. PMID: 30389665 Free PMC article.
-
Slit diaphragm dysfunction in proteinuric states: identification of novel therapeutic targets for nephrotic syndrome.Clin Exp Nephrol. 2009 Aug;13(4):275-280. doi: 10.1007/s10157-009-0162-x. Epub 2009 Mar 7. Clin Exp Nephrol. 2009. PMID: 19266252 Review.
-
ER-associated degradation in health and disease - from substrate to organism.J Cell Sci. 2019 Dec 2;132(23):jcs232850. doi: 10.1242/jcs.232850. J Cell Sci. 2019. PMID: 31792042 Free PMC article. Review.
Cited by
-
The mechanisms to dispose of misfolded proteins in the endoplasmic reticulum of adipocytes.Nat Commun. 2023 May 30;14(1):3132. doi: 10.1038/s41467-023-38690-4. Nat Commun. 2023. PMID: 37253728 Free PMC article.
-
Endoplasmic Reticulum-Associated Protein Degradation.Cold Spring Harb Perspect Biol. 2022 Dec 1;14(12):a041247. doi: 10.1101/cshperspect.a041247. Cold Spring Harb Perspect Biol. 2022. PMID: 35940909 Free PMC article. Review.
-
Disease predictability review using common biomarkers appearing in diabetic nephropathy and neurodegeneration of experimental animals.Lab Anim Res. 2022 Feb 7;38(1):3. doi: 10.1186/s42826-022-00113-8. Lab Anim Res. 2022. PMID: 35130988 Free PMC article. Review.
-
Glycyrrhizic Acid Protects Glomerular Podocytes Induced by High Glucose by Modulating SNARK/AMPK Signaling Pathway.Curr Med Sci. 2023 Aug;43(4):696-707. doi: 10.1007/s11596-023-2765-y. Epub 2023 Jul 14. Curr Med Sci. 2023. PMID: 37450070
-
SEL1L-mediated endoplasmic reticulum associated degradation inhibition suppresses proliferation and migration in Huh7 hepatocellular carcinoma cells.World J Gastroenterol. 2025 Mar 14;31(10):103133. doi: 10.3748/wjg.v31.i10.103133. World J Gastroenterol. 2025. PMID: 40093667 Free PMC article.
References
-
- Garg P. A review of podocyte biology. Am J Nephrol. 2018;47(suppl 1):3–13. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
