

Excessive O-GlcNAcylation Causes Heart Failure and Sudden Death
- PMID: 33593071
- PMCID: PMC8085112
- DOI: 10.1161/CIRCULATIONAHA.120.051911
Excessive O-GlcNAcylation Causes Heart Failure and Sudden Death
Erratum in
-
Correction to: Excessive O-GlcNAcylation Causes Heart Failure and Sudden Death.Circulation. 2021 Apr 27;143(17):e892. doi: 10.1161/CIR.0000000000000976. Epub 2021 Apr 26. Circulation. 2021. PMID: 33900835 No abstract available.
Abstract
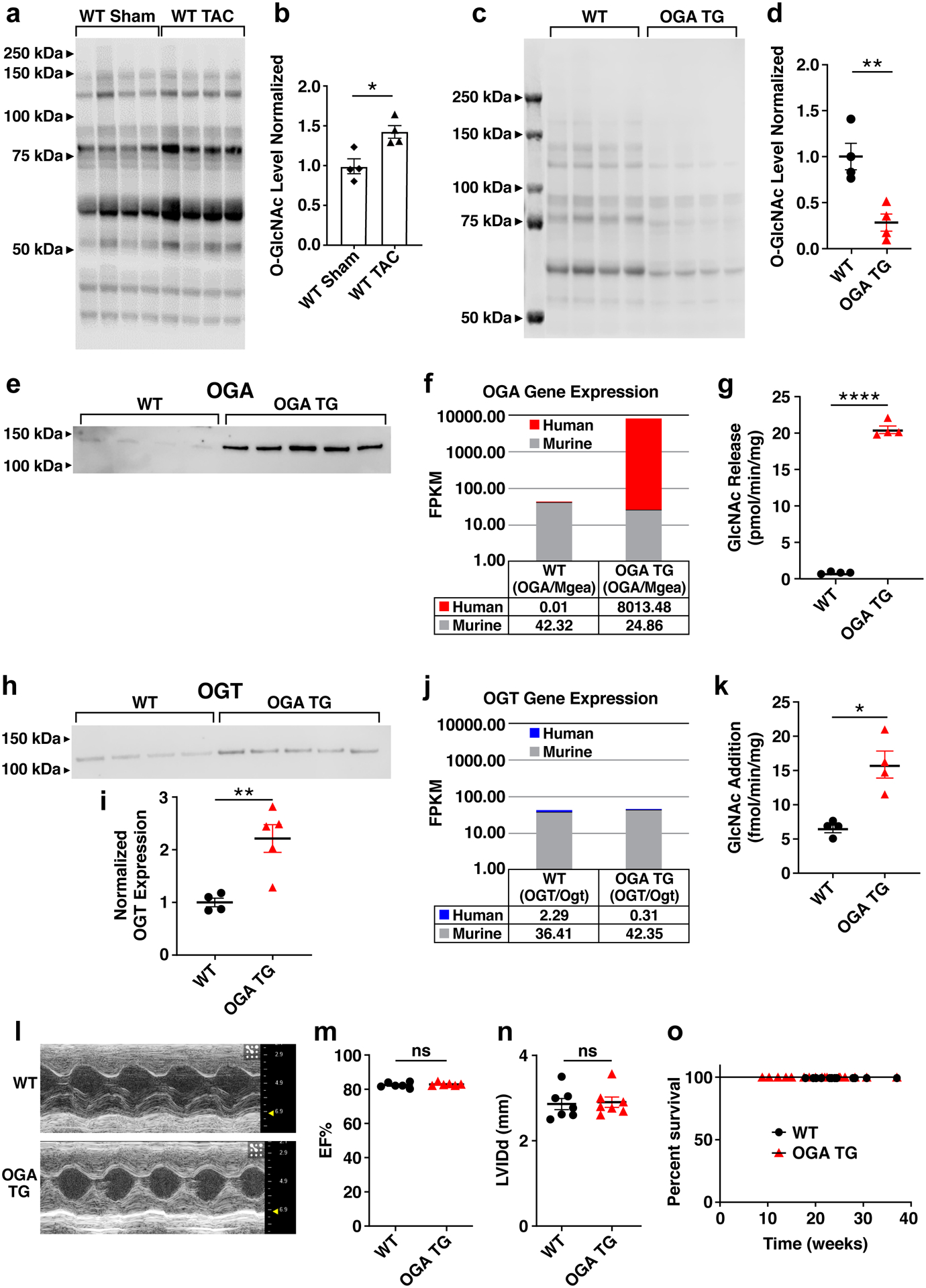
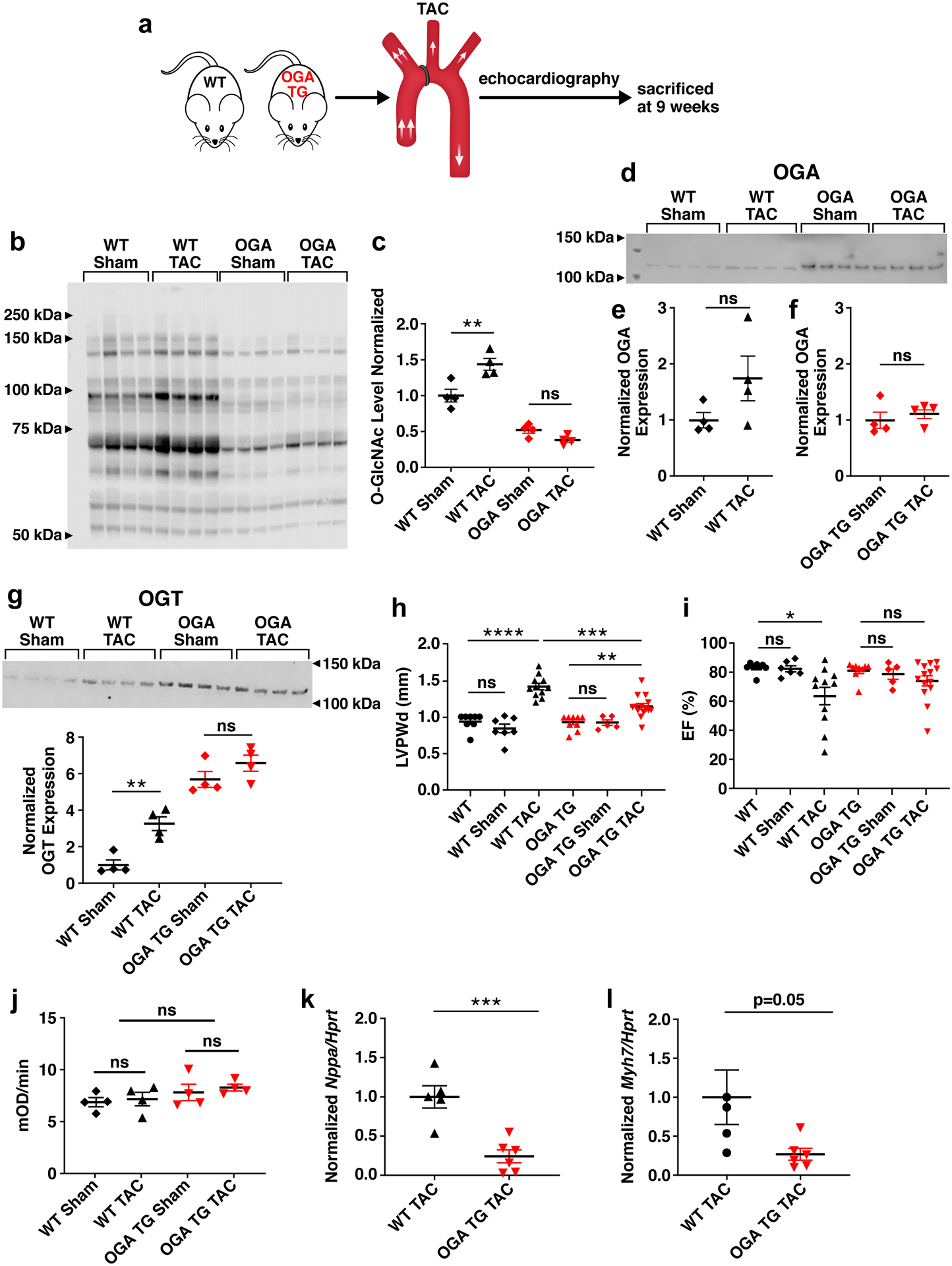
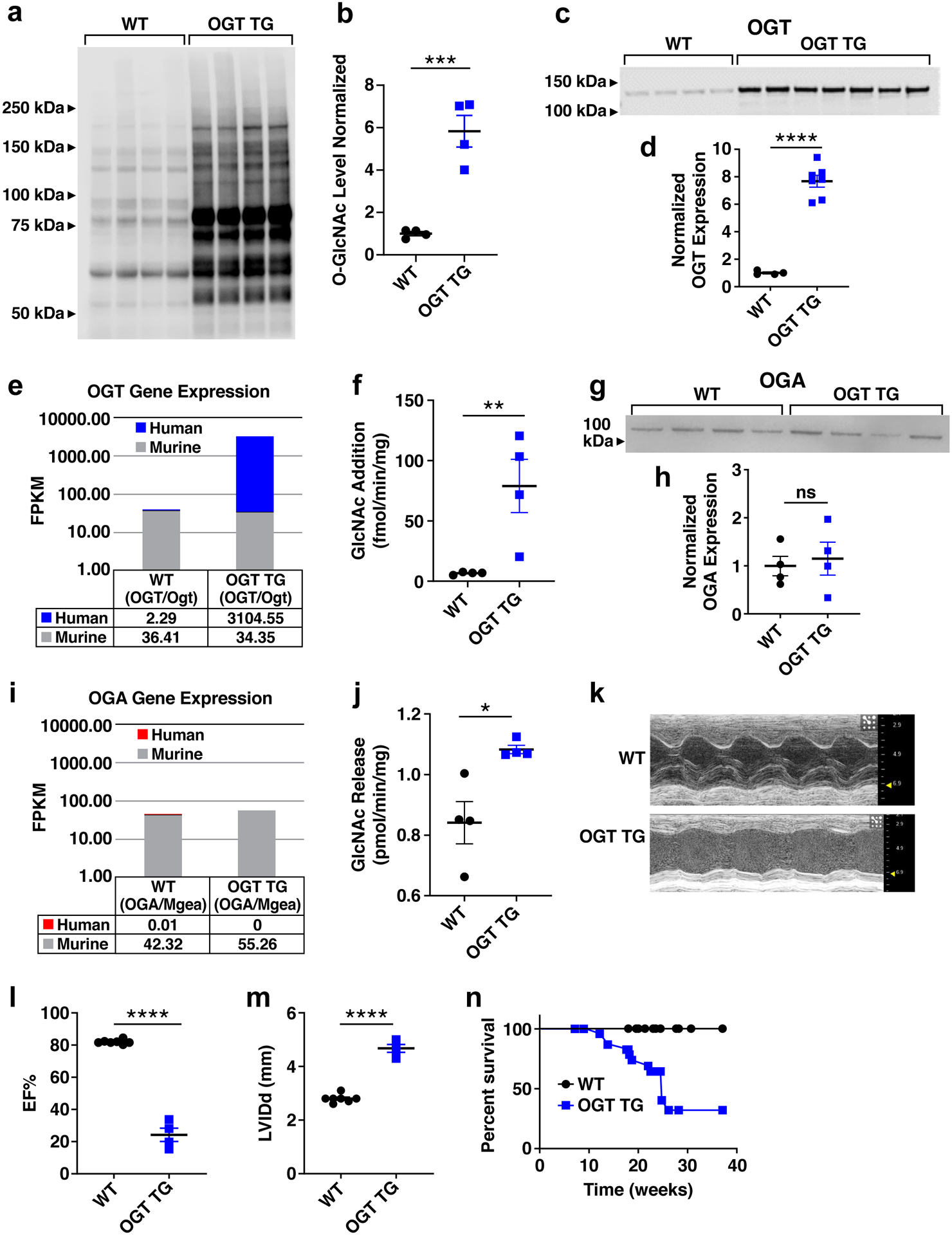
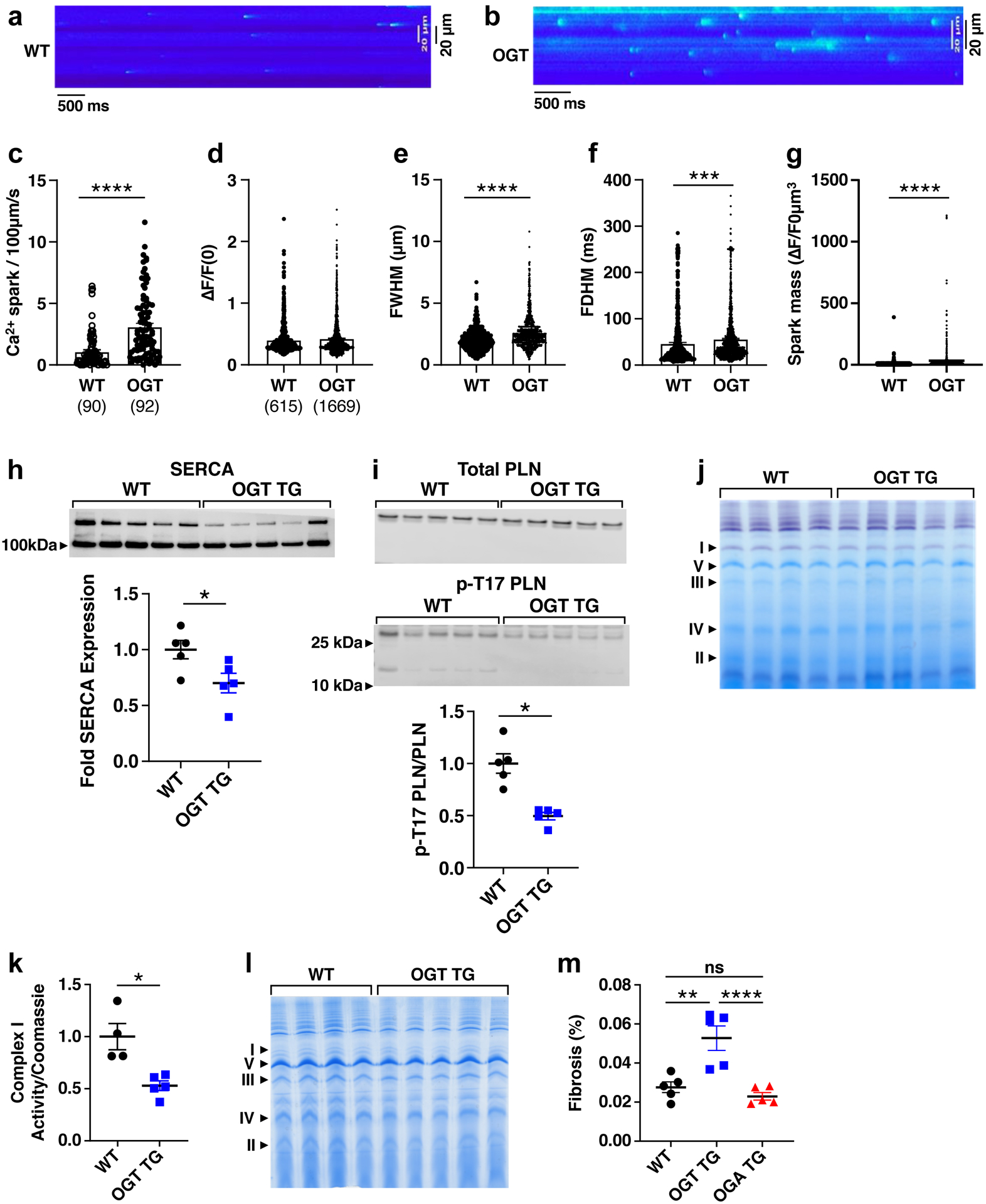
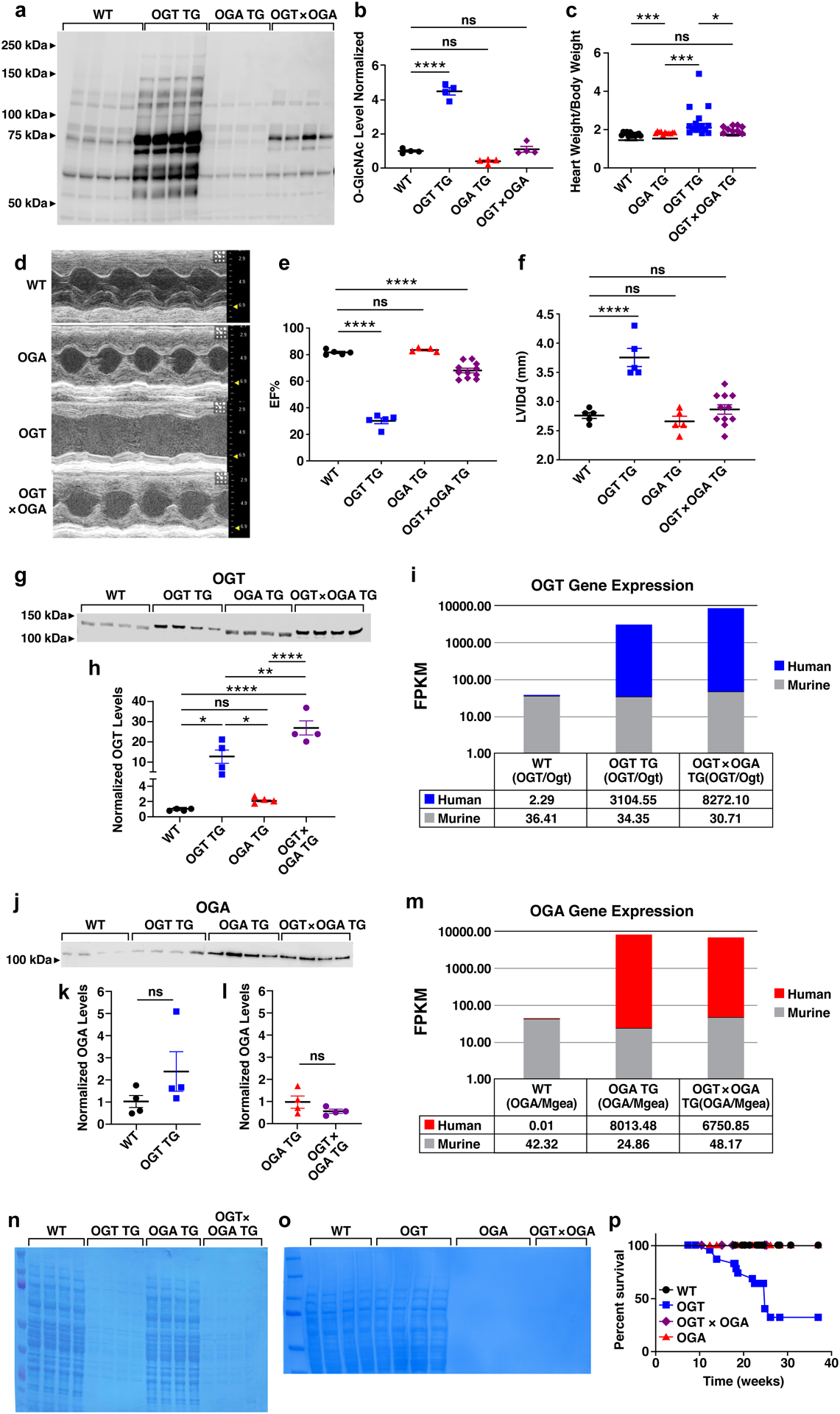
Background: Heart failure is a leading cause of death worldwide and is associated with the rising prevalence of obesity, hypertension, and diabetes. O-GlcNAcylation (the attachment of O-linked β-N-acetylglucosamine [O-GlcNAc] moieties to cytoplasmic, nuclear, and mitochondrial proteins) is a posttranslational modification of intracellular proteins and serves as a metabolic rheostat for cellular stress. Total levels of O-GlcNAcylation are determined by nutrient and metabolic flux, in addition to the net activity of 2 enzymes: O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA). Failing myocardium is marked by increased O-GlcNAcylation, but whether excessive O-GlcNAcylation contributes to cardiomyopathy and heart failure is unknown.
Methods: We developed 2 new transgenic mouse models with myocardial overexpression of OGT and OGA to control O-GlcNAcylation independent of pathologic stress.
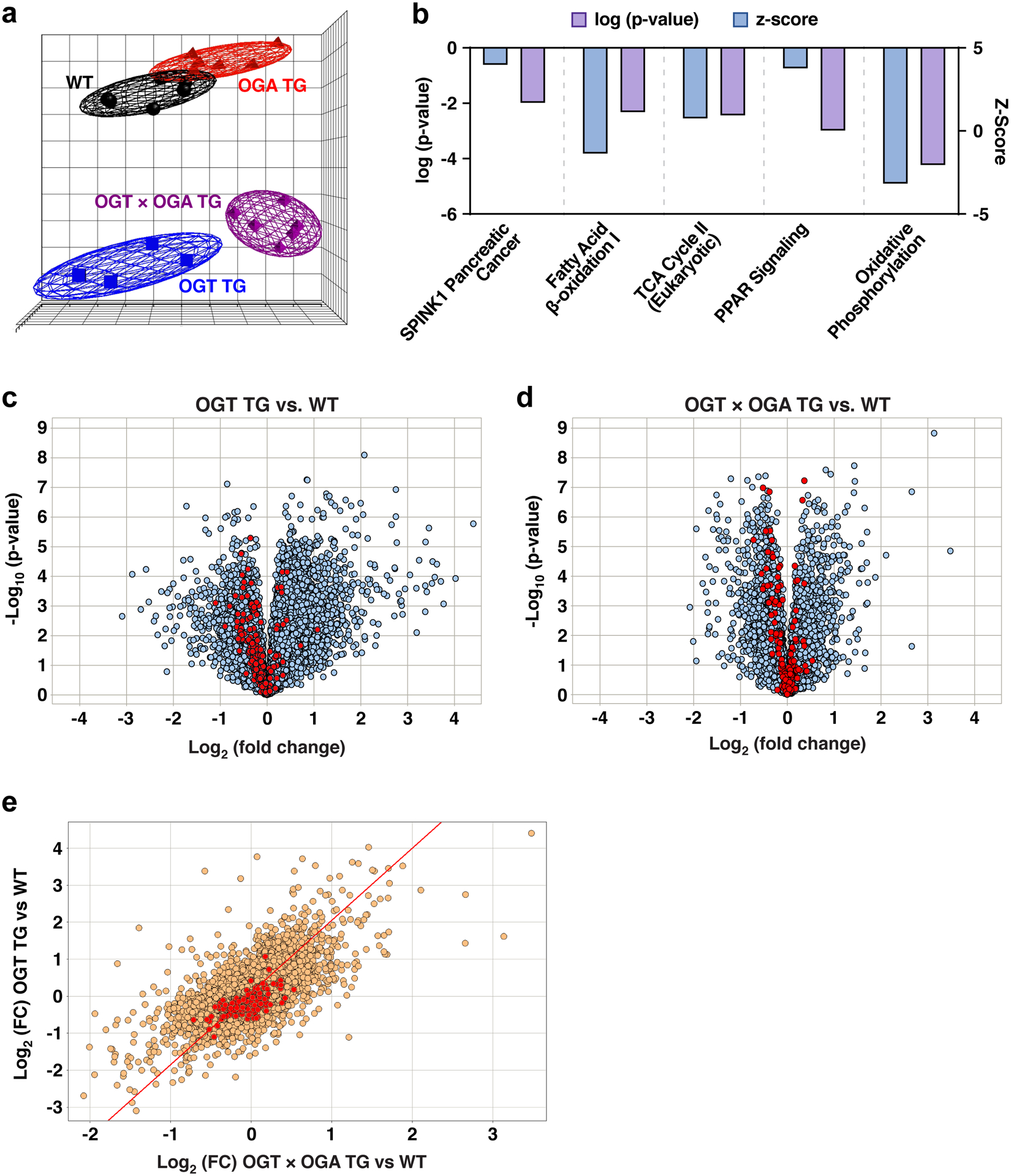
Results: We found that OGT transgenic hearts showed increased O-GlcNAcylation and developed severe dilated cardiomyopathy, ventricular arrhythmias, and premature death. In contrast, OGA transgenic hearts had lower O-GlcNAcylation but identical cardiac function to wild-type littermate controls. OGA transgenic hearts were resistant to pathologic stress induced by pressure overload with attenuated myocardial O-GlcNAcylation levels after stress and decreased pathologic hypertrophy compared with wild-type controls. Interbreeding OGT with OGA transgenic mice rescued cardiomyopathy and premature death, despite persistent elevation of myocardial OGT. Transcriptomic and functional studies revealed disrupted mitochondrial energetics with impairment of complex I activity in hearts from OGT transgenic mice. Complex I activity was rescued by OGA transgenic interbreeding, suggesting an important role for mitochondrial complex I in O-GlcNAc-mediated cardiac pathology.
Conclusions: Our data provide evidence that excessive O-GlcNAcylation causes cardiomyopathy, at least in part, attributable to defective energetics. Enhanced OGA activity is well tolerated and attenuation of O-GlcNAcylation is beneficial against pressure overload-induced pathologic remodeling and heart failure. These findings suggest that attenuation of excessive O-GlcNAcylation may represent a novel therapeutic approach for cardiomyopathy.
Keywords: CaMKII; Uridine diphosphate N-acetylglucosamine; cardiac arrhythmia; heart failure; hypertension; hypertrophy; post-translational protein processing.
Figures

References
-
- Ida GL, Jan Magnus A, Heidi K, Eirik Q, Ivar S, Theis T, Geir C, Line MG-W and Cathrine RC. Cardiac O-GlcNAc signaling is increased in hypertrophy and heart failure. Physiological Genomics. 2012;44:162–172. - PubMed
-
- Lehmann LH, Jebessa ZH, Kreusser MM, Horsch A, He T, Kronlage M, Dewenter M, Sramek V, Oehl U, Krebs-Haupenthal J, von der Lieth AH, Schmidt A, Sun Q, Ritterhoff J, Finke D, Volkers M, Jungmann A, Sauer SW, Thiel C, Nickel A, Kohlhaas M, Schafer M, Sticht C, Maack C, Gretz N, Wagner M, El-Armouche A, Maier LS, Londono JEC, Meder B, Freichel M, Grone HJ, Most P, Muller OJ, Herzig S, Furlong EEM, Katus HA and Backs J. A proteolytic fragment of histone deacetylase 4 protects the heart from failure by regulating the hexosamine biosynthetic pathway. Nat Med. 2018;24:62–72. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
