

Mutations in PRDM15 Are a Novel Cause of Galloway-Mowat Syndrome
- PMID: 33593823
- PMCID: PMC7920168
- DOI: 10.1681/ASN.2020040490
Mutations in PRDM15 Are a Novel Cause of Galloway-Mowat Syndrome
Abstract
Background: Galloway-Mowat syndrome (GAMOS) is characterized by neurodevelopmental defects and a progressive nephropathy, which typically manifests as steroid-resistant nephrotic syndrome. The prognosis of GAMOS is poor, and the majority of children progress to renal failure. The discovery of monogenic causes of GAMOS has uncovered molecular pathways involved in the pathogenesis of disease.
Methods: Homozygosity mapping, whole-exome sequencing, and linkage analysis were used to identify mutations in four families with a GAMOS-like phenotype, and high-throughput PCR technology was applied to 91 individuals with GAMOS and 816 individuals with isolated nephrotic syndrome. In vitro and in vivo studies determined the functional significance of the mutations identified.
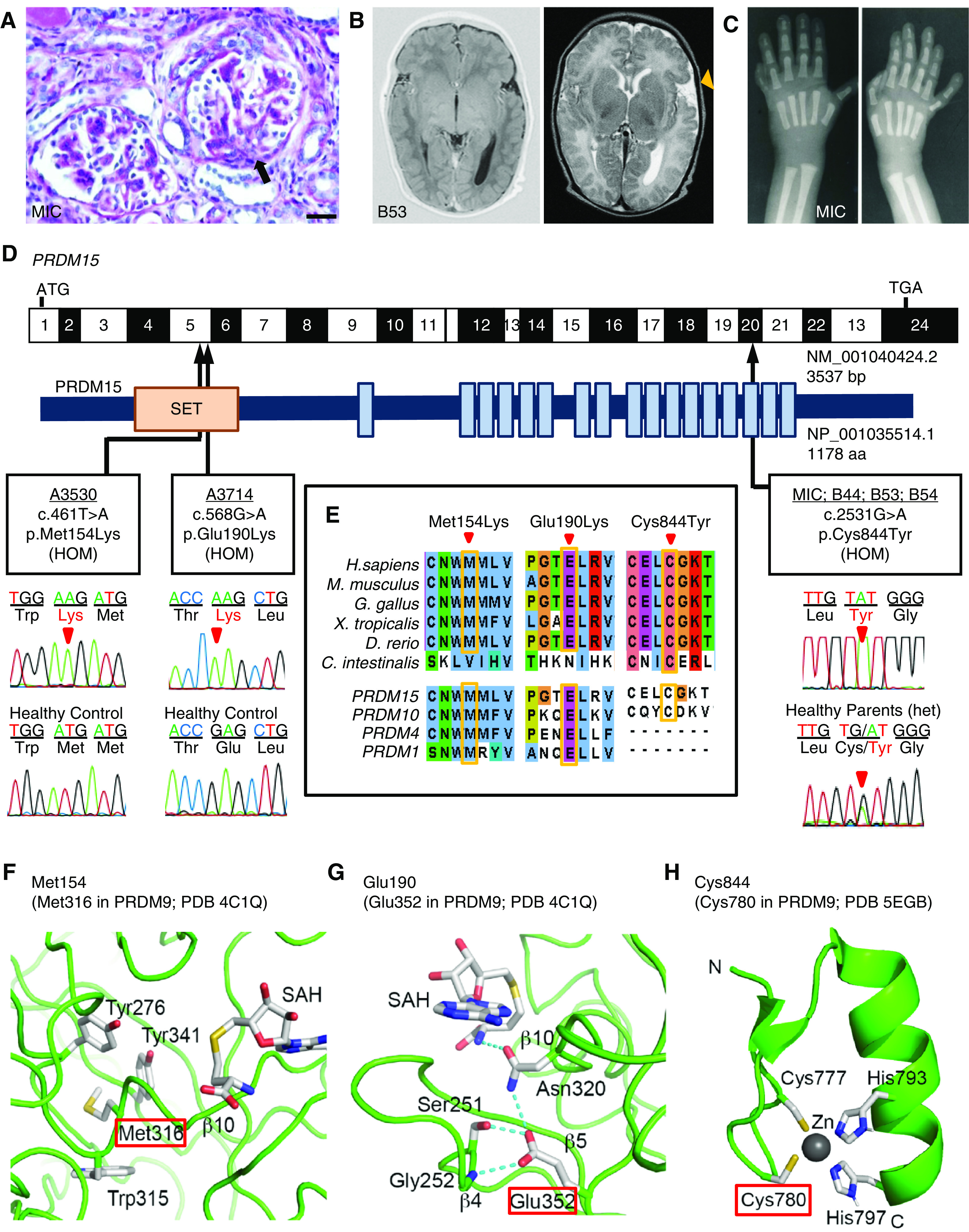
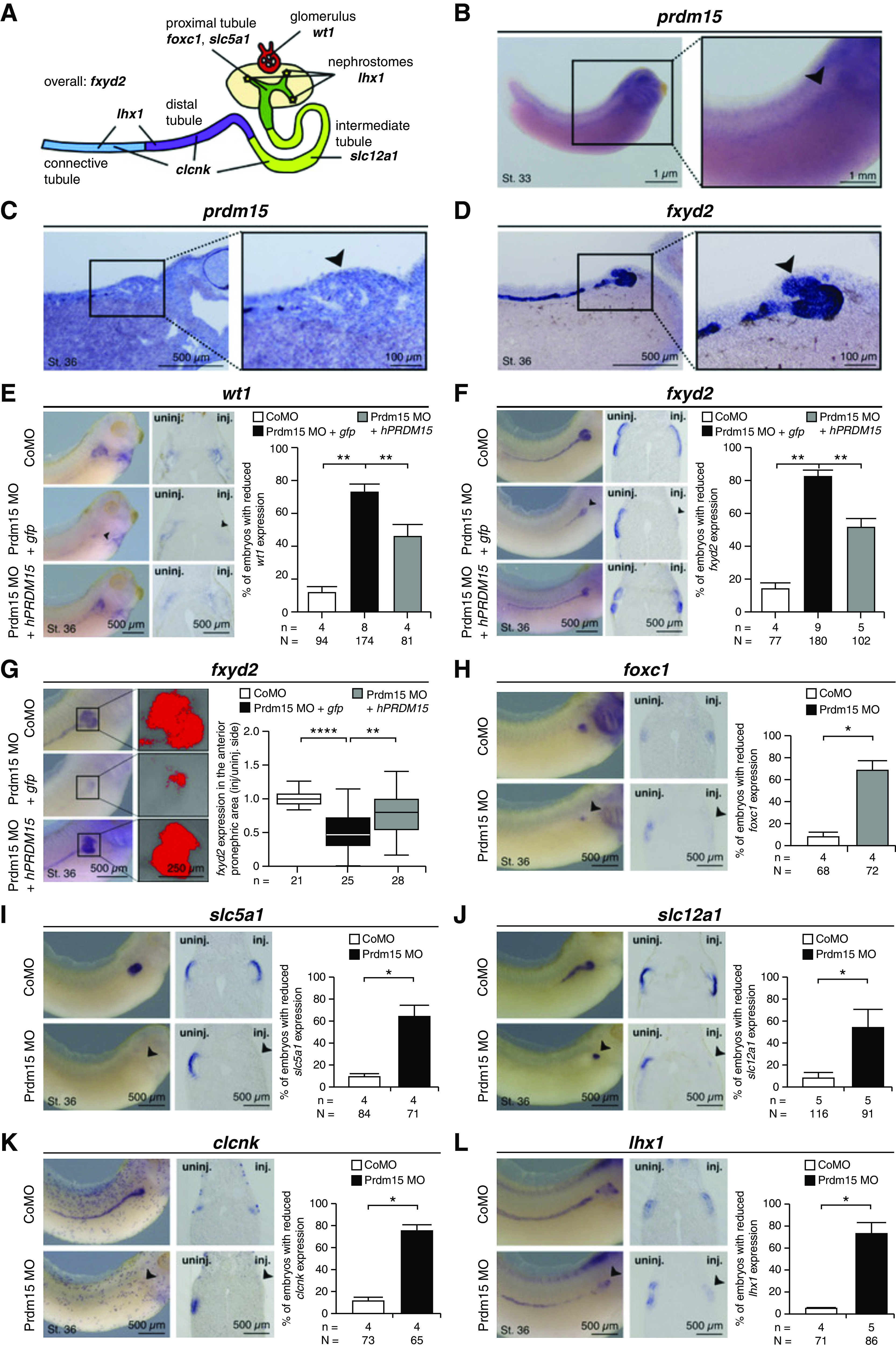
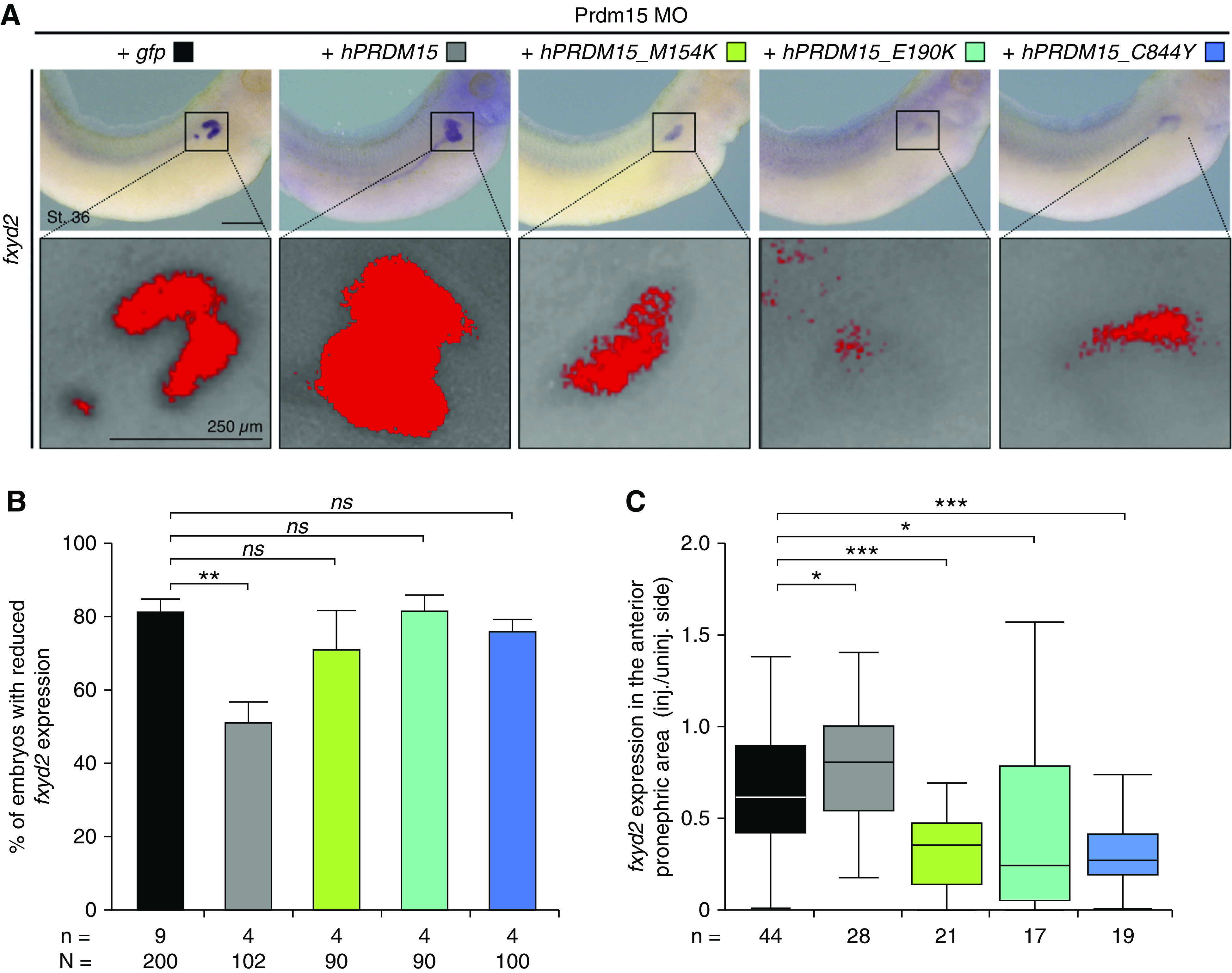
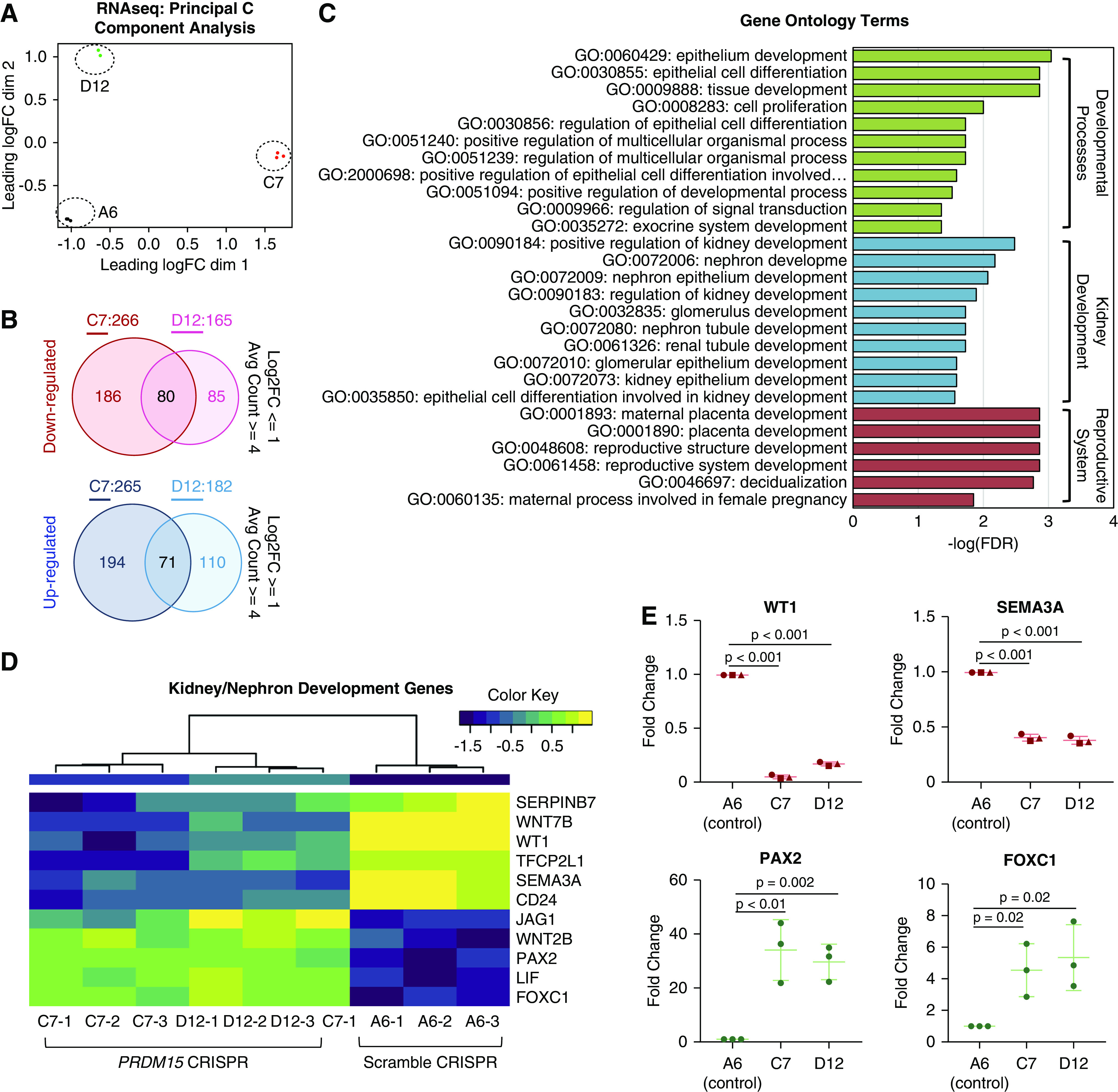
Results: Three biallelic variants of the transcriptional regulator PRDM15 were detected in six families with proteinuric kidney disease. Four families with a variant in the protein's zinc-finger (ZNF) domain have additional GAMOS-like features, including brain anomalies, cardiac defects, and skeletal defects. All variants destabilize the PRDM15 protein, and the ZNF variant additionally interferes with transcriptional activation. Morpholino oligonucleotide-mediated knockdown of Prdm15 in Xenopus embryos disrupted pronephric development. Human wild-type PRDM15 RNA rescued the disruption, but the three PRDM15 variants did not. Finally, CRISPR-mediated knockout of PRDM15 in human podocytes led to dysregulation of several renal developmental genes.
Conclusions: Variants in PRDM15 can cause either isolated nephrotic syndrome or a GAMOS-type syndrome on an allelic basis. PRDM15 regulates multiple developmental kidney genes, and is likely to play an essential role in renal development in humans.
Keywords: genetic renal disease; genetics and development; nephrotic syndrome.
Copyright © 2021 by the American Society of Nephrology.
Figures


Similar articles
-
Homozygous splicing mutation in NUP133 causes Galloway-Mowat syndrome.Ann Neurol. 2018 Dec;84(6):814-828. doi: 10.1002/ana.25370. Ann Neurol. 2018. PMID: 30427554
-
Homozygous mutation in NUP107 leads to microcephaly with steroid-resistant nephrotic condition similar to Galloway-Mowat syndrome.J Med Genet. 2017 Jun;54(6):399-403. doi: 10.1136/jmedgenet-2016-104237. Epub 2017 Mar 9. J Med Genet. 2017. PMID: 28280135
-
Mutations in WDR4 as a new cause of Galloway-Mowat syndrome.Am J Med Genet A. 2018 Nov;176(11):2460-2465. doi: 10.1002/ajmg.a.40489. Epub 2018 Aug 6. Am J Med Genet A. 2018. PMID: 30079490 Free PMC article.
-
Novel homozygous OSGEP gene pathogenic variants in two unrelated patients with Galloway-Mowat syndrome: case report and review of the literature.BMC Nephrol. 2019 Apr 11;20(1):126. doi: 10.1186/s12882-019-1317-y. BMC Nephrol. 2019. PMID: 30975089 Free PMC article. Review.
-
Genetics and phenotypic heterogeneity of Galloway-Mowat syndrome.Cell Commun Signal. 2025 Jun 18;23(1):289. doi: 10.1186/s12964-025-02307-8. Cell Commun Signal. 2025. PMID: 40533795 Free PMC article. Review.
Cited by
-
Galloway-Mowat Syndrome Type 3 Caused by OSGEP Gene Variants: A Case Report and Literature Review.Front Pediatr. 2022 Jun 17;10:899991. doi: 10.3389/fped.2022.899991. eCollection 2022. Front Pediatr. 2022. PMID: 35783322 Free PMC article.
-
Prdm15 acts upstream of Wnt4 signaling in anterior neural development of Xenopus laevis.Front Cell Dev Biol. 2024 Feb 20;12:1316048. doi: 10.3389/fcell.2024.1316048. eCollection 2024. Front Cell Dev Biol. 2024. PMID: 38444828 Free PMC article.
-
Xenopus laevis (Daudin, 1802) as a Model Organism for Bioscience: A Historic Review and Perspective.Biology (Basel). 2023 Jun 20;12(6):890. doi: 10.3390/biology12060890. Biology (Basel). 2023. PMID: 37372174 Free PMC article. Review.
-
WHAMM functions in kidney reabsorption and polymerizes actin to promote autophagosomal membrane closure and cargo sequestration.bioRxiv [Preprint]. 2024 Jan 23:2024.01.22.576497. doi: 10.1101/2024.01.22.576497. bioRxiv. 2024. Update in: Mol Biol Cell. 2024 Jun 1;35(6):ar80. doi: 10.1091/mbc.E24-01-0025. PMID: 38328079 Free PMC article. Updated. Preprint.
-
Case Report: Novel compound heterozygous TPRKB variants cause Galloway-Mowat syndrome.Front Pediatr. 2024 Apr 3;12:1360867. doi: 10.3389/fped.2024.1360867. eCollection 2024. Front Pediatr. 2024. PMID: 38628357 Free PMC article.
References
-
- Cohen AH, Turner MC: Kidney in galloway-mowat syndrome: Clinical spectrum with description of pathology. Kidney Int 45: 1407–1415, 1994. 10.1038/ki.1994.184 - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
