

Experimentally Validated Reconstruction and Analysis of a Genome-Scale Metabolic Model of an Anaerobic Neocallimastigomycota Fungus
- PMID: 33594000
- PMCID: PMC8561657
- DOI: 10.1128/mSystems.00002-21
Experimentally Validated Reconstruction and Analysis of a Genome-Scale Metabolic Model of an Anaerobic Neocallimastigomycota Fungus
Abstract
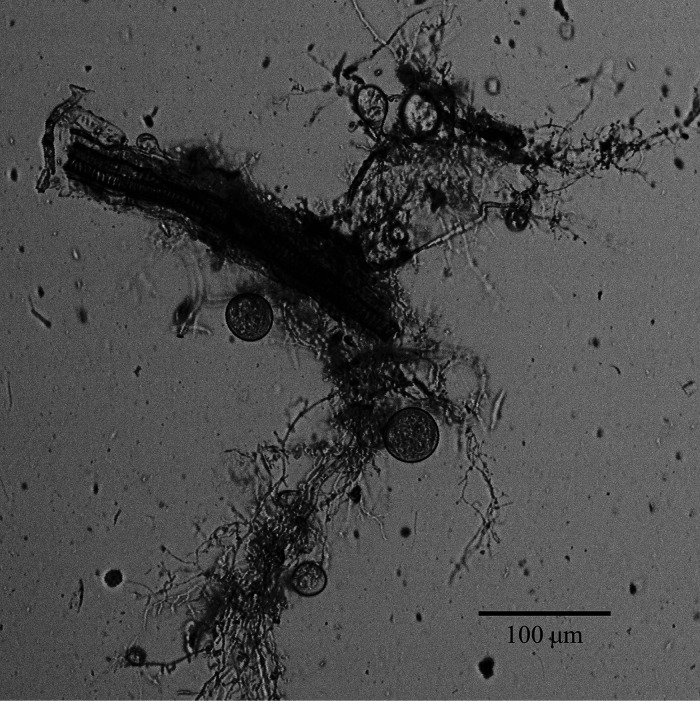
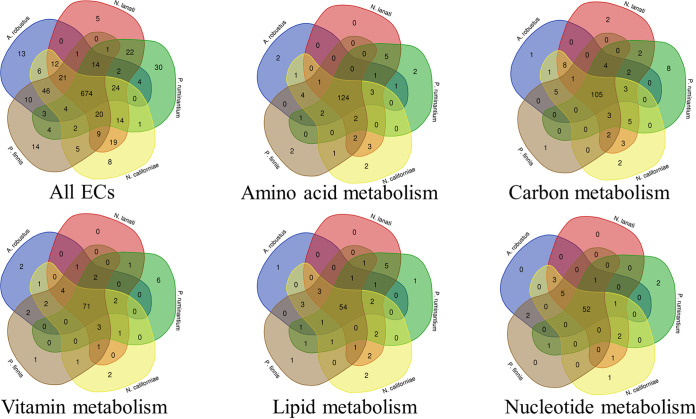
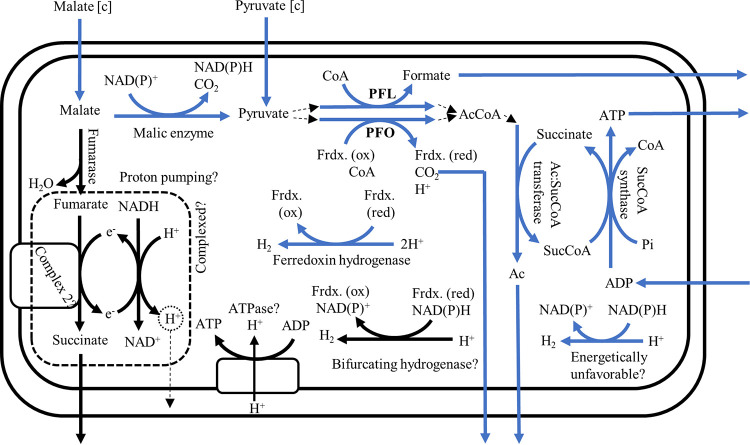
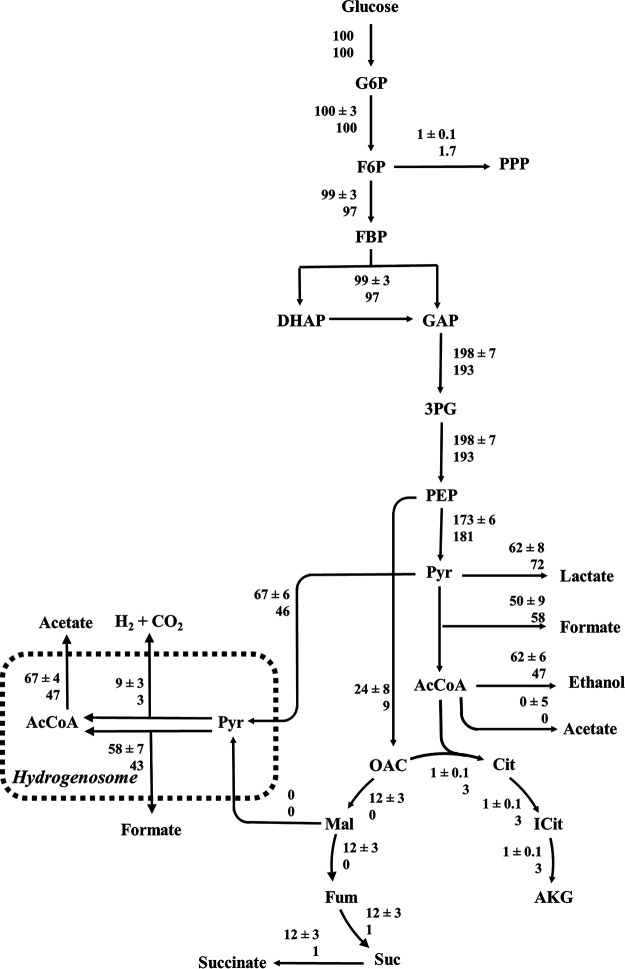
Anaerobic gut fungi in the phylum Neocallimastigomycota typically inhabit the digestive tracts of large mammalian herbivores, where they play an integral role in the decomposition of raw lignocellulose into its constitutive sugar monomers. However, quantitative tools to study their physiology are lacking, partially due to their complex and unresolved metabolism that includes the largely uncharacterized fungal hydrogenosome. Modern omics approaches combined with metabolic modeling can be used to establish an understanding of gut fungal metabolism and develop targeted engineering strategies to harness their degradation capabilities for lignocellulosic bioprocessing. Here, we introduce a high-quality genome of the anaerobic fungus Neocallimastix lanati from which we constructed the first genome-scale metabolic model of an anaerobic fungus. Relative to its size (200 Mbp, sequenced at 62× depth), it is the least fragmented publicly available gut fungal genome to date. Of the 1,788 lignocellulolytic enzymes annotated in the genome, 585 are associated with the fungal cellulosome, underscoring the powerful lignocellulolytic potential of N. lanati The genome-scale metabolic model captures the primary metabolism of N. lanati and accurately predicts experimentally validated substrate utilization requirements. Additionally, metabolic flux predictions are verified by 13C metabolic flux analysis, demonstrating that the model faithfully describes the underlying fungal metabolism. Furthermore, the model clarifies key aspects of the hydrogenosomal metabolism and can be used as a platform to quantitatively study these biotechnologically important yet poorly understood early-branching fungi.IMPORTANCE Recent genomic analyses have revealed that anaerobic gut fungi possess both the largest number and highest diversity of lignocellulolytic enzymes of all sequenced fungi, explaining their ability to decompose lignocellulosic substrates, e.g., agricultural waste, into fermentable sugars. Despite their potential, the development of engineering methods for these organisms has been slow due to their complex life cycle, understudied metabolism, and challenging anaerobic culture requirements. Currently, there is no framework that can be used to combine multi-omic data sets to understand their physiology. Here, we introduce a high-quality PacBio-sequenced genome of the anaerobic gut fungus Neocallimastix lanati Beyond identifying a trove of lignocellulolytic enzymes, we use this genome to construct the first genome-scale metabolic model of an anaerobic gut fungus. The model is experimentally validated and sheds light on unresolved metabolic features common to gut fungi. Model-guided analysis will pave the way for deepening our understanding of anaerobic gut fungi and provides a systematic framework to guide strain engineering efforts of these organisms for biotechnological use.
Keywords: 13C metabolic flux analysis; Neocallimastigomycota; Neocallimastix lanati; anaerobes; anaerobic fungi; flux balance analysis; genome-scale metabolic model; nonmodel fungus.
Copyright © 2021 Wilken et al.
Figures
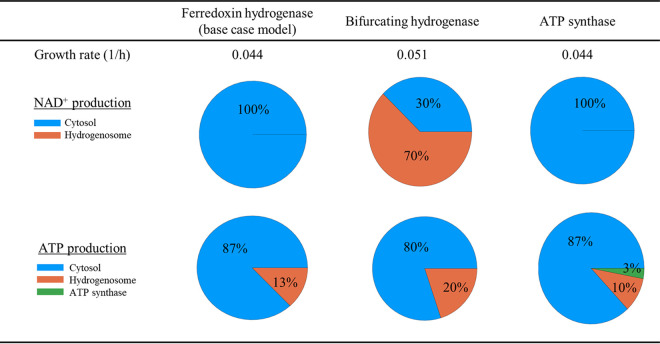
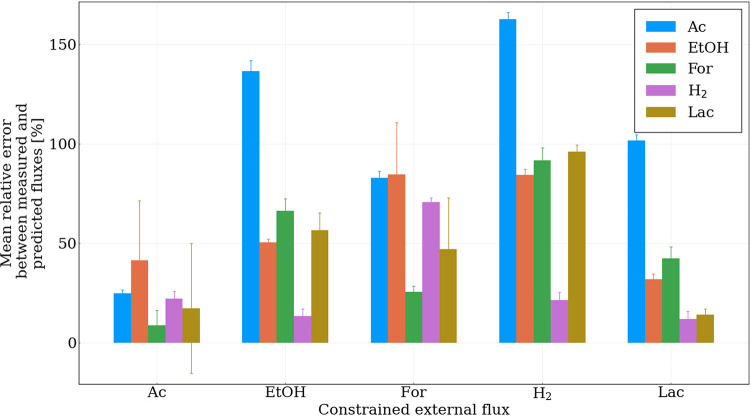
References
-
- Gruninger RJ, Puniya AK, Callaghan TM, Edwards JE, Youssef N, Dagar SS, Fliegerova K, Griffith GW, Forster R, Tsang A, Mcallister T, Elshahed MS. 2014. Anaerobic fungi (phylum Neocallimastigomycota): advances in understanding their taxonomy, life cycle, ecology, role and biotechnological potential. FEMS Microbiol Ecol 90:1–17. doi:10.1111/1574-6941.12383. - DOI - PubMed
-
- Hackstein JHP, Baker SE, van Hellemond JJ, Tielens AGM. 2019. Hydrogenosomes of anaerobic fungi: an alternative way to adapt to anaerobic environments, p 159–175. Springer, Cham, Switzerland.
-
- Solomon KV, Haitjema CH, Henske JK, Gilmore SP, Borges-Rivera D, Lipzen A, Brewer HM, Purvine SO, Wright AT, Theodorou MK, Grigoriev IV, Regev A, Thompson DA, O'Malley MA. 2016. Early-branching gut fungi possess a large, comprehensive array of biomass-degrading enzymes. Science 351:1192–1196. doi:10.1126/science.aad1431. - DOI - PMC - PubMed
-
- Haitjema CH, Gilmore SP, Henske JK, Solomon KV, de Groot R, Kuo A, Mondo SJ, Salamov AA, LaButti K, Zhao Z, Chiniquy J, Barry K, Brewer HM, Purvine SO, Wright AT, Hainaut M, Boxma B, van Alen T, Hackstein JHP, Henrissat B, Baker SE, Grigoriev IV, O'Malley MA. 2017. A parts list for fungal cellulosomes revealed by comparative genomics. Nat Microbiol 2:17087. doi:10.1038/nmicrobiol.2017.87. - DOI - PubMed
-
- Resch MG, Donohoe BS, Baker JO, Decker SR, Bayer EA, Beckham GT, Himmel ME. 2013. Fungal cellulases and complexed cellulosomal enzymes exhibit synergistic mechanisms in cellulose deconstruction. Energy Environ Sci 6:1858–1867. doi:10.1039/c3ee00019b. - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
