

Analysis of metagenome-assembled viral genomes from the human gut reveals diverse putative CrAss-like phages with unique genomic features
- PMID: 33594055
- PMCID: PMC7886860
- DOI: 10.1038/s41467-021-21350-w
Analysis of metagenome-assembled viral genomes from the human gut reveals diverse putative CrAss-like phages with unique genomic features
Abstract
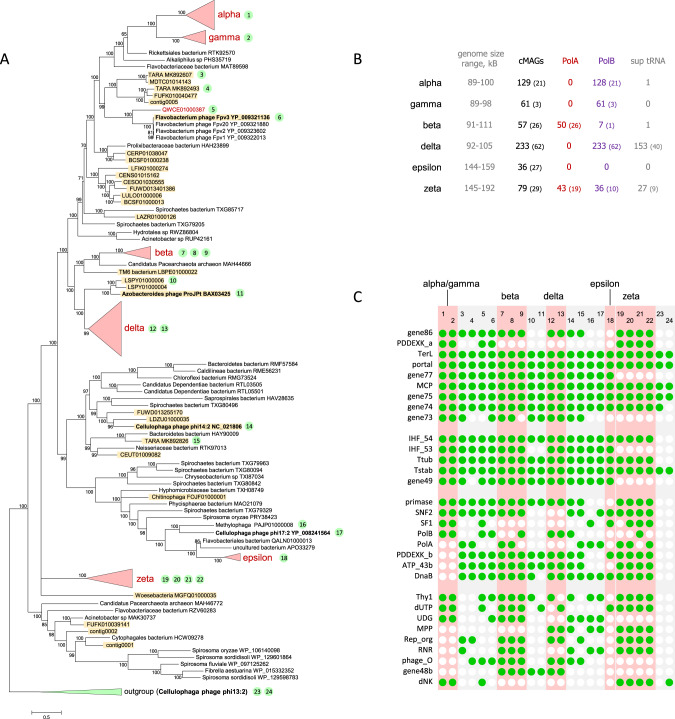
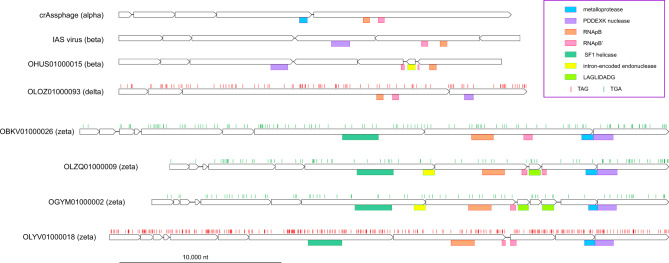
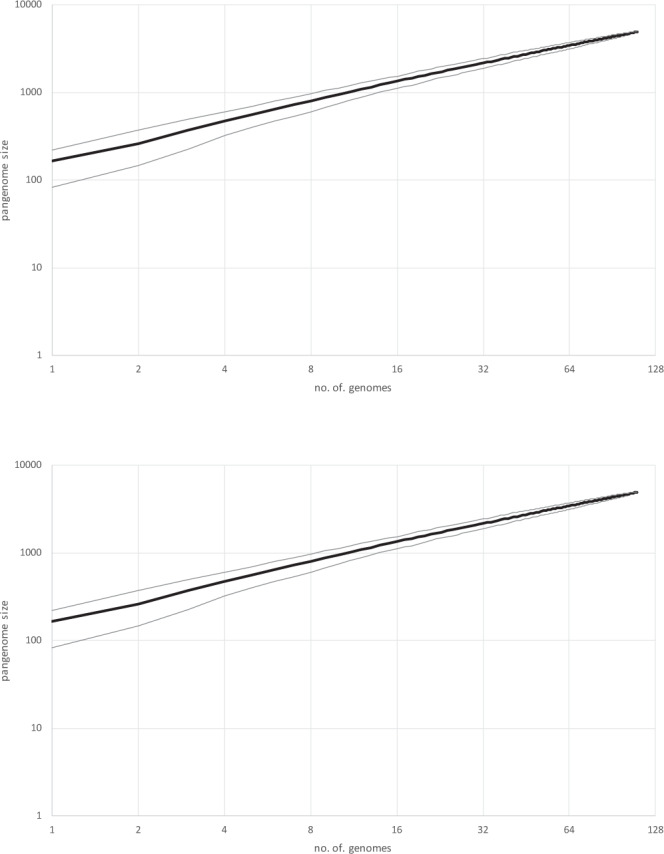
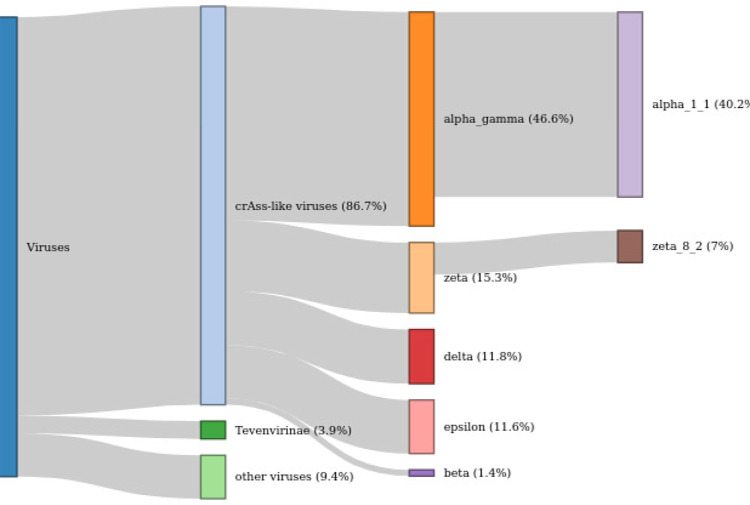
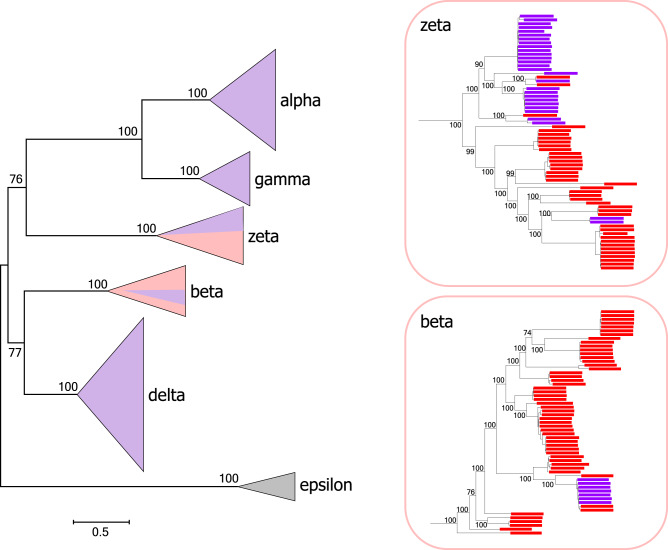
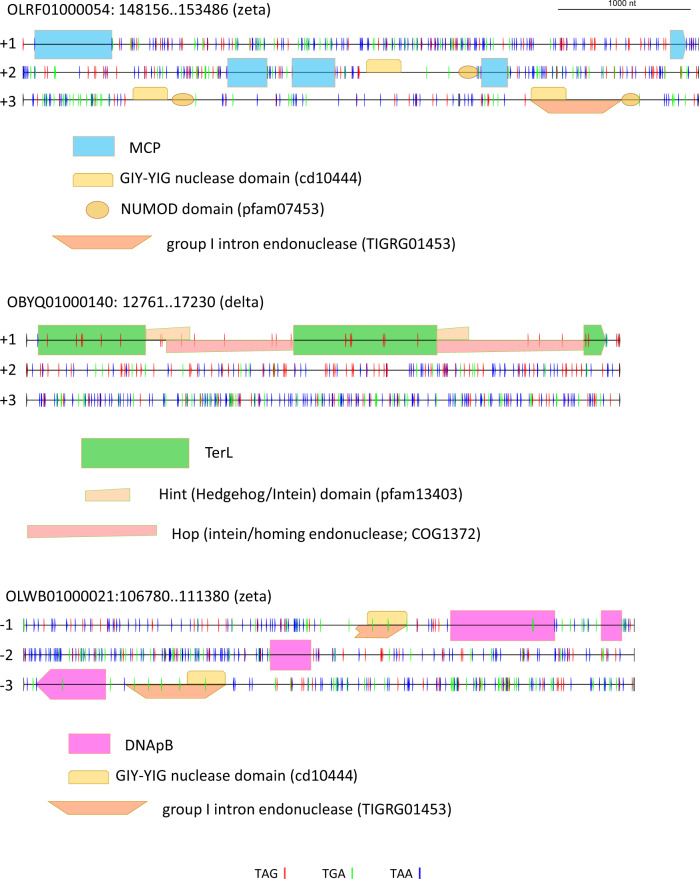
CrAssphage is the most abundant human-associated virus and the founding member of a large group of bacteriophages, discovered in animal-associated and environmental metagenomes, that infect bacteria of the phylum Bacteroidetes. We analyze 4907 Circular Metagenome Assembled Genomes (cMAGs) of putative viruses from human gut microbiomes and identify nearly 600 genomes of crAss-like phages that account for nearly 87% of the DNA reads mapped to these cMAGs. Phylogenetic analysis of conserved genes demonstrates the monophyly of crAss-like phages, a putative virus order, and of 5 branches, potential families within that order, two of which have not been identified previously. The phage genomes in one of these families are almost twofold larger than the crAssphage genome (145-192 kilobases), with high density of self-splicing introns and inteins. Many crAss-like phages encode suppressor tRNAs that enable read-through of UGA or UAG stop-codons, mostly, in late phage genes. A distinct feature of the crAss-like phages is the recurrent switch of the phage DNA polymerase type between A and B families. Thus, comparative genomic analysis of the expanded assemblage of crAss-like phages reveals aspects of genome architecture and expression as well as phage biology that were not apparent from the previous work on phage genomics.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Hurwitz, B. L., U’Ren, J. M. & Youens-Clark, K. Computational prospecting the great viral unknown. FEMS Microbiol. Lett. 10.1093/femsle/fnw077 (2016). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
