

This is a preprint.
SARS-CoV-2 European resurgence foretold: interplay of introductions and persistence by leveraging genomic and mobility data
- PMID: 33594355
- PMCID: PMC7885927
- DOI: 10.21203/rs.3.rs-208849/v1
SARS-CoV-2 European resurgence foretold: interplay of introductions and persistence by leveraging genomic and mobility data
Update in
-
Untangling introductions and persistence in COVID-19 resurgence in Europe.Nature. 2021 Jul;595(7869):713-717. doi: 10.1038/s41586-021-03754-2. Epub 2021 Jun 30. Nature. 2021. PMID: 34192736 Free PMC article.
Abstract
Following the first wave of SARS-CoV-2 infections in spring 2020, Europe experienced a resurgence of the virus starting late summer that was deadlier and more difficult to contain. Relaxed intervention measures and summer travel have been implicated as drivers of the second wave. Here, we build a phylogeographic model to evaluate how newly introduced lineages, as opposed to the rekindling of persistent lineages, contributed to the COVID-19 resurgence in Europe. We inform this model using genomic, mobility and epidemiological data from 10 West European countries and estimate that in many countries more than 50% of the lineages circulating in late summer resulted from new introductions since June 15th. The success in onwards transmission of these lineages is predicted by SARS-CoV-2 incidence during this period. Relatively early introductions from Spain into the United Kingdom contributed to the successful spread of the 20A.EU1/B.1.177 variant. The pervasive spread of variants that have not been associated with an advantage in transmissibility highlights the threat of novel variants of concern that emerged more recently and have been disseminated by holiday travel. Our findings indicate that more effective and coordinated measures are required to contain spread through cross-border travel.
Conflict of interest statement
Competing Interests
The authors declare no competing interests.
Figures

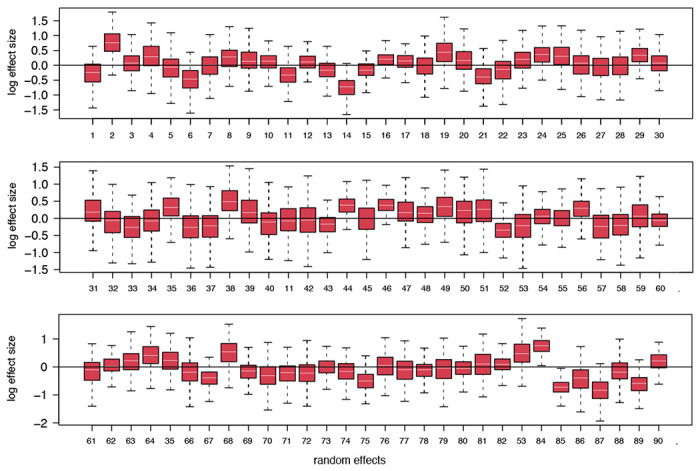
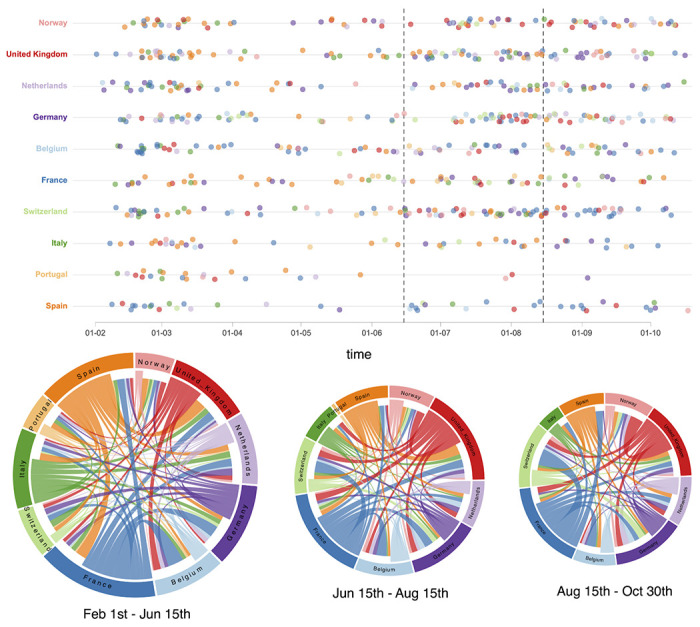
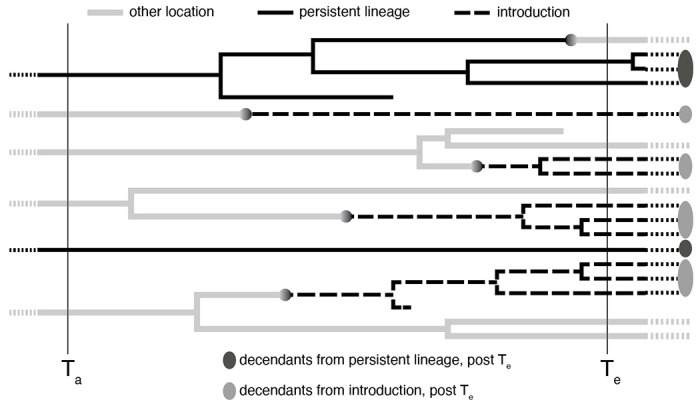



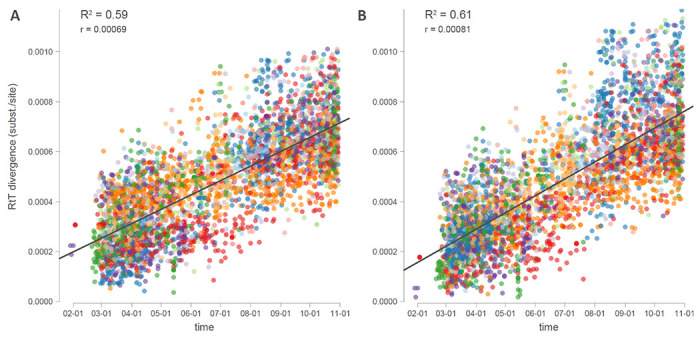
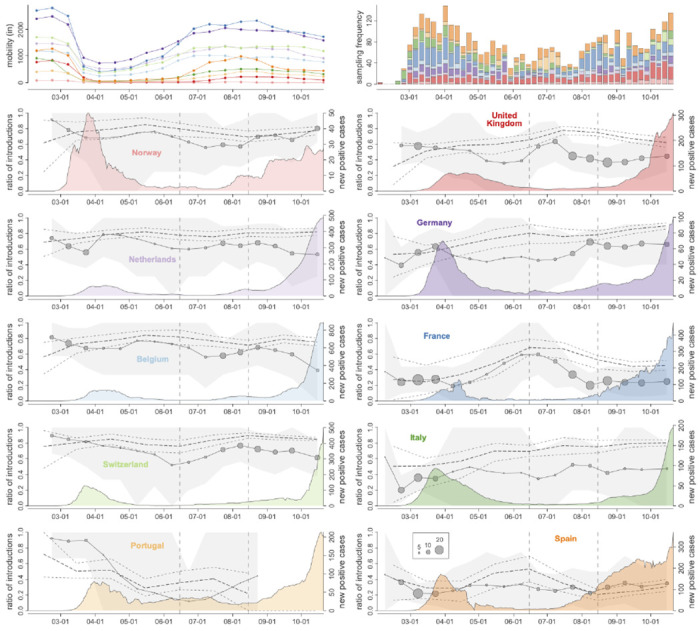

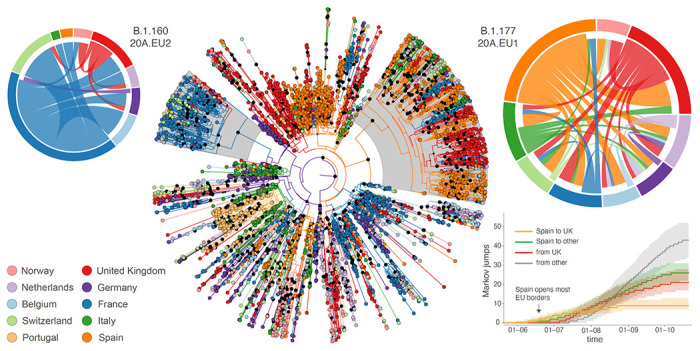
References
-
- Riley S. et al. High prevalence of SARS-CoV-2 swab positivity and increasing R number in England during October 2020: REACT-1 round 6 interim report. bioRxiv (2020) doi: 10.1101/2020.10.30.20223123 - DOI
-
- Data on 14-day notification rate of new COVID-19 cases and deaths. https://www.ecdc.europa.eu/en/publications-data/data-national-14-day-not... (2021).
-
- COVID-19 situation update for the EU/EEA, as of week 3, updated 28 January 2021 https://www.ecdc.europa.eu/en/cases-2019-ncov-eueea.
-
- Rambaut Andrew, Loman Nick, Pybus Oliver, Barclay Wendy, Barrett Jeff, Carabelli Alesandro, Connor Tom, Peacock Tom, Robertson David L, Volz Erik, on behalf of COVID-19 Genomics Consortium UK (CoG-UK). Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations, virological.orghttps://virological.org/t/preliminary-genomic-characterisation-of-an-eme... (2020).
-
- Volz E. et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. bioRxiv (2021) doi: 10.1101/2020.12.30.20249034 - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
