

Cardiac amyloidosis-A review of current literature for the practicing physician
- PMID: 33595871
- PMCID: PMC7943900
- DOI: 10.1002/clc.23572
Cardiac amyloidosis-A review of current literature for the practicing physician
Abstract
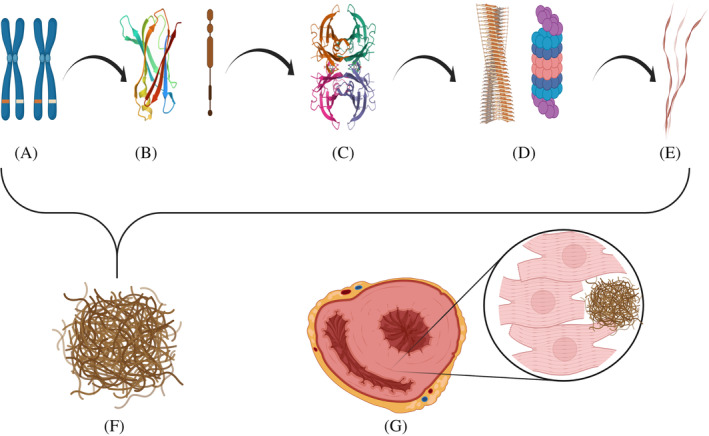
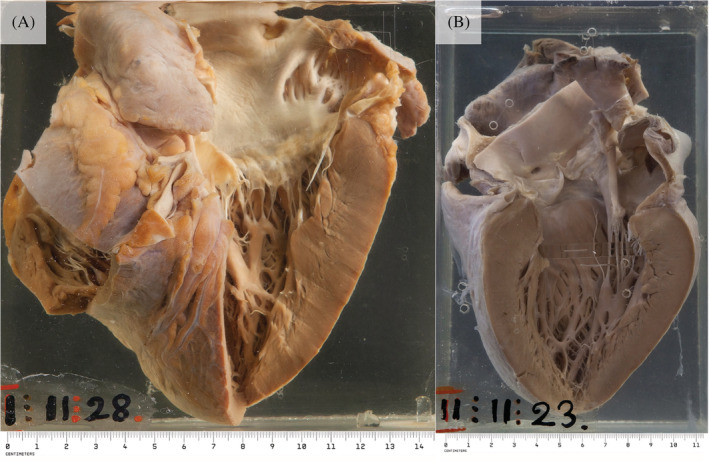
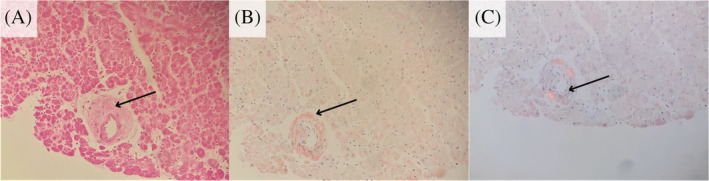
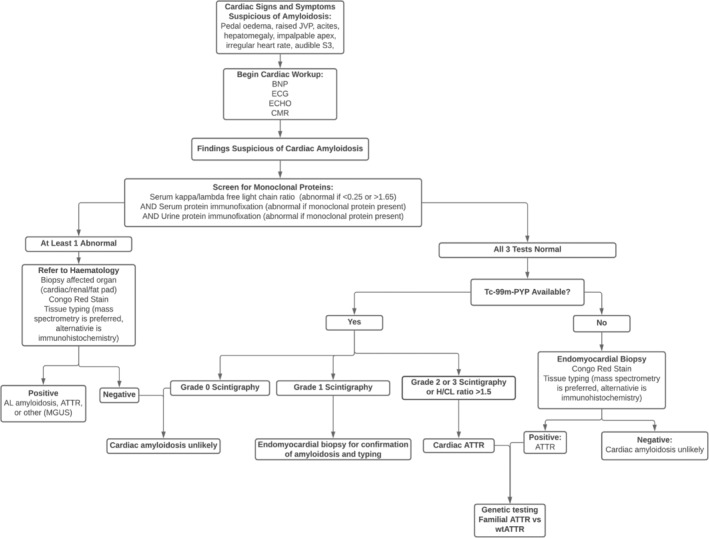
The amyloidoses are a family of diseases in which misfolded precursor proteins aggregate to form amyloid and deposit in body tissues. A very serious yet underrecognized form of this disease is cardiac amyloidosis, in which amyloid deposits into the extracellular space of the myocardium, resulting in thickening and stiffening of ventricular walls with resultant heart failure and conductive dysfunction. This review provides a discussion of the pathogenesis and clinical presentation of cardiac amyloidosis subtypes, as well as an up-to-date approach to diagnosis and treatment. Significant progress has been made in recent years regarding diagnosis and treatment of this condition, but prognosis remains heavily reliant on early detection of the disease. Two types of precursor protein are responsible for most cardiac amyloidosis cases: transthyretin amyloid, and immunoglobulin-derived light chain amyloid. An early diagnosis of cardiac amyloidosis can allow for novel treatment modalities to be initiated with the potential to improve prognosis.
Keywords: AL; ATTR; amyloid deposits; amyloidosis; cardiac amyloidosis.
© 2021 The Authors. Clinical Cardiology published by Wiley Periodicals LLC.
Conflict of interest statement
None.
Figures
References
-
- Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97:75‐84. - PubMed
-
- Kittleson M, Maurer M, Ambardekar A, et al. Cardiac amyloidosis: evolving diagnosis and management: a scientific statement from the American Heart Association. Circulation. 2020;142(1):e7‐e22. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
