

Mapping major SARS-CoV-2 drug targets and assessment of druggability using computational fragment screening: Identification of an allosteric small-molecule binding site on the Nsp13 helicase
- PMID: 33596235
- PMCID: PMC7888625
- DOI: 10.1371/journal.pone.0246181
Mapping major SARS-CoV-2 drug targets and assessment of druggability using computational fragment screening: Identification of an allosteric small-molecule binding site on the Nsp13 helicase
Abstract
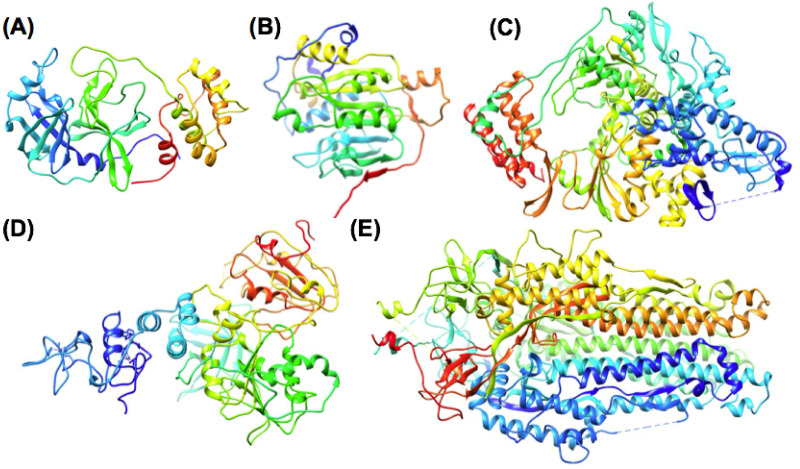
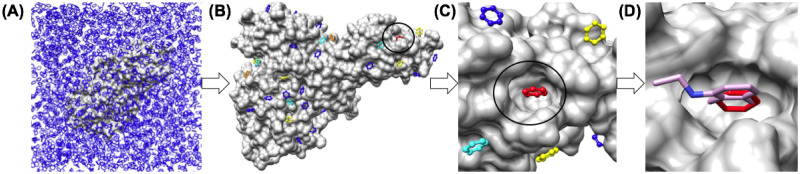
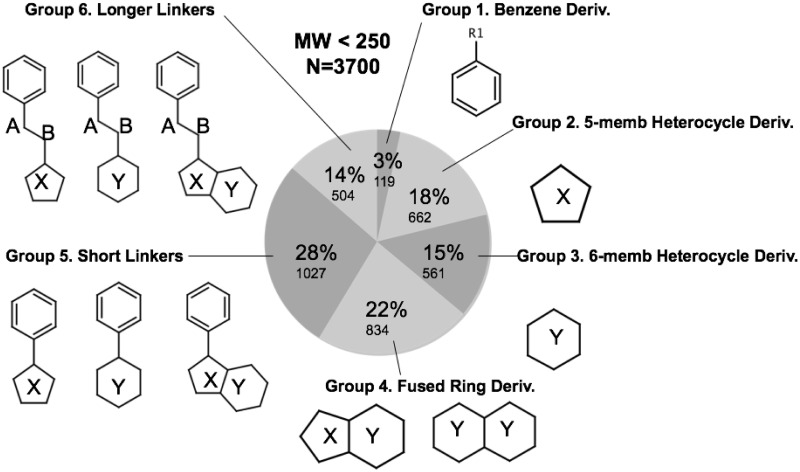
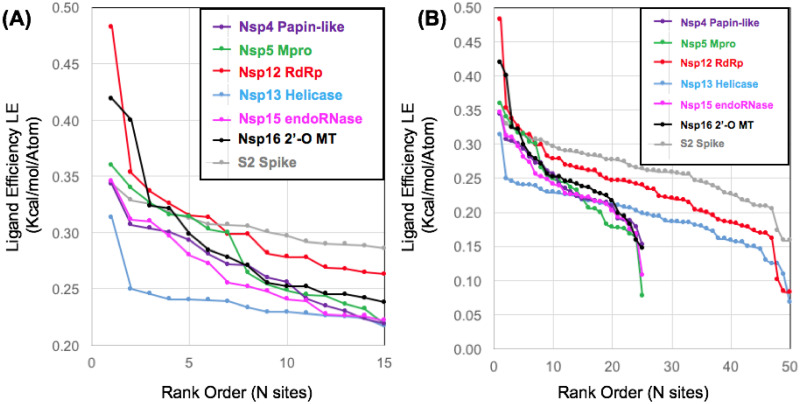
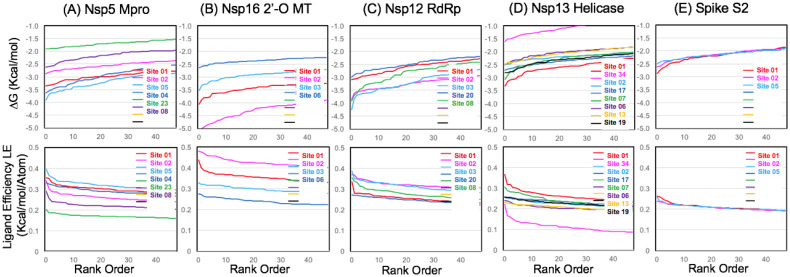
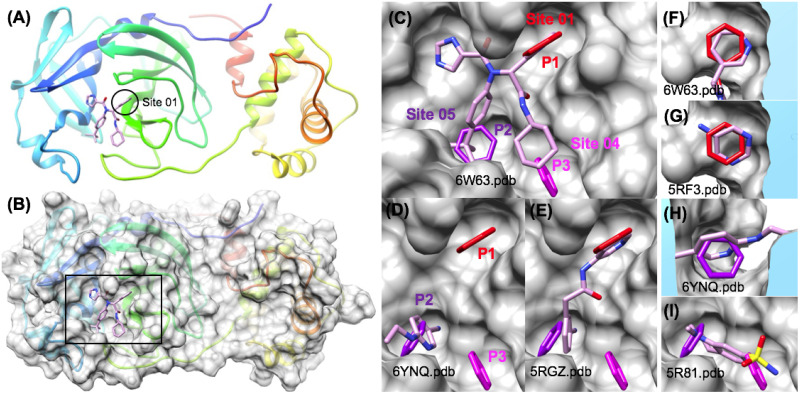
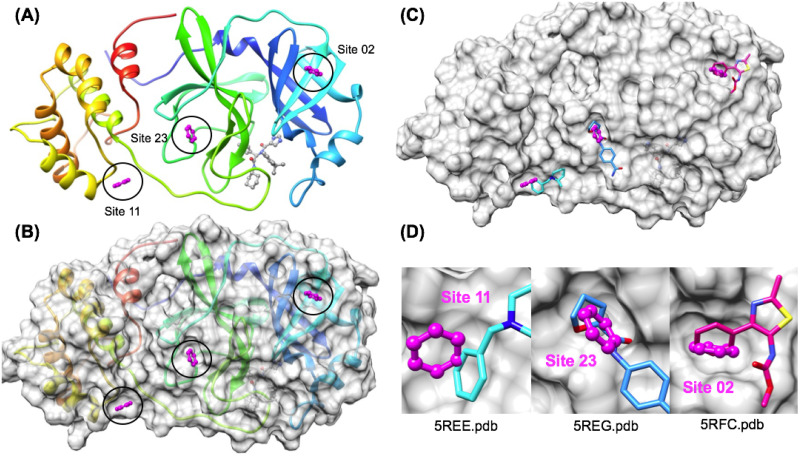
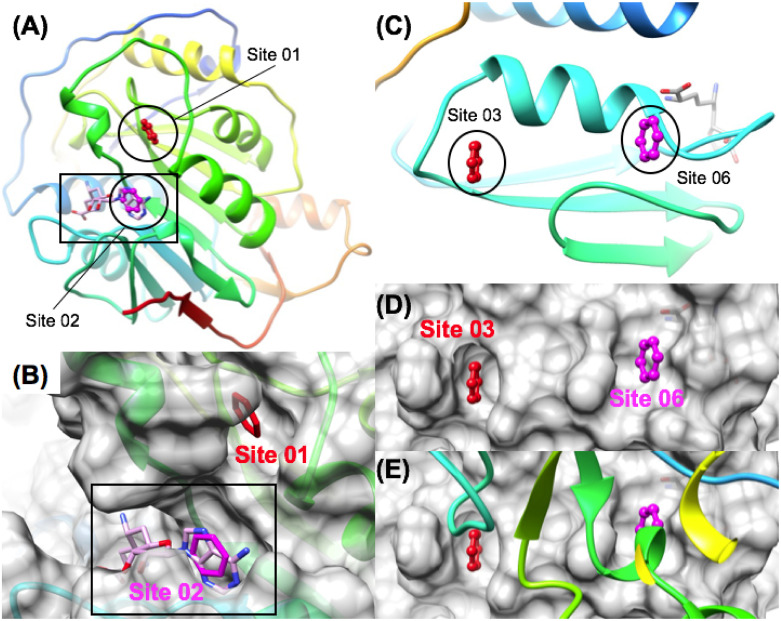
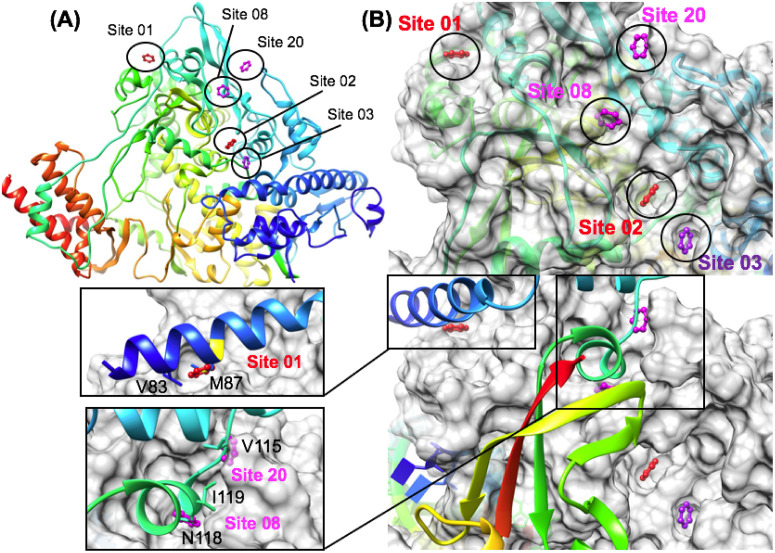
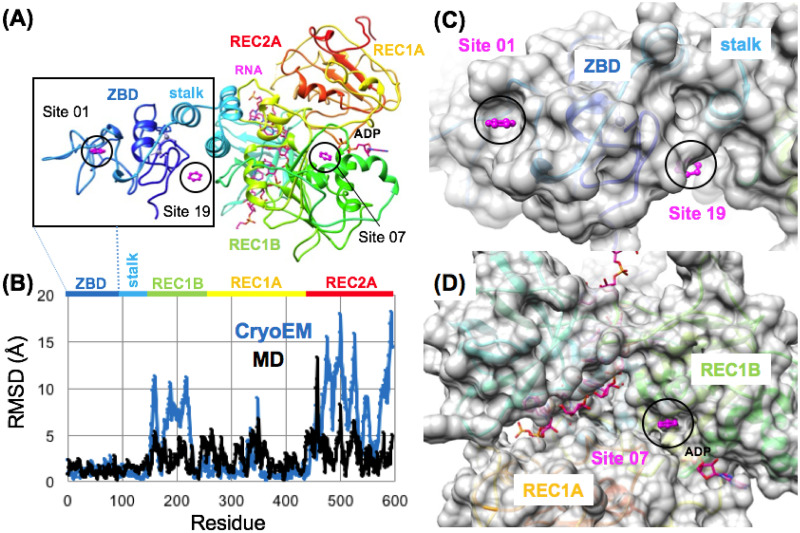
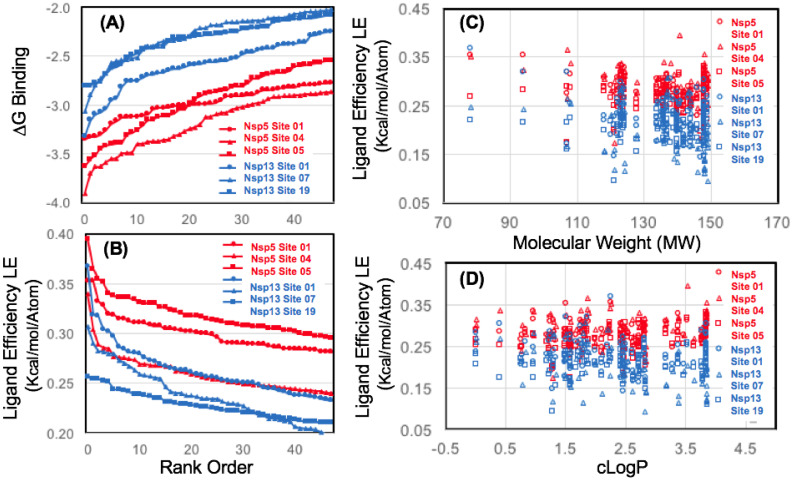
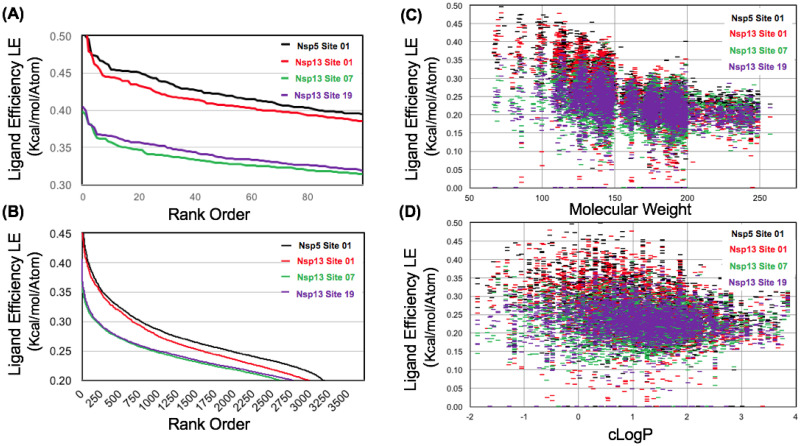
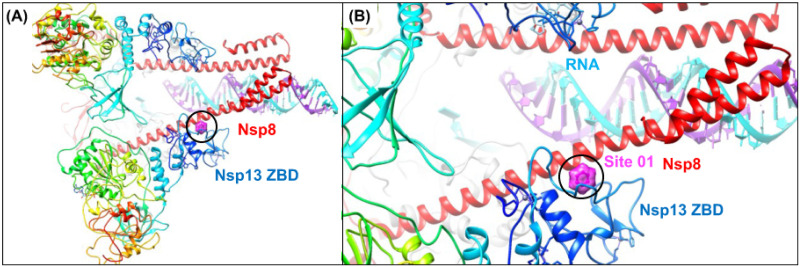
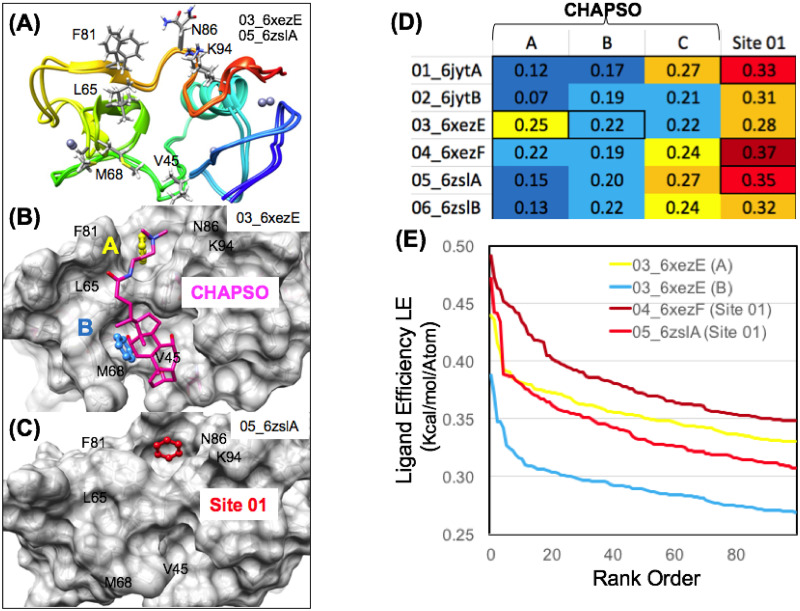
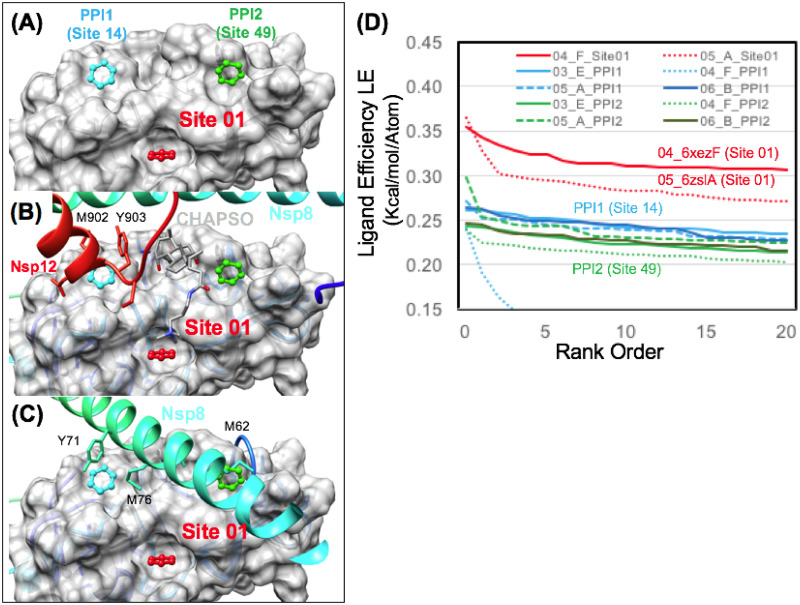
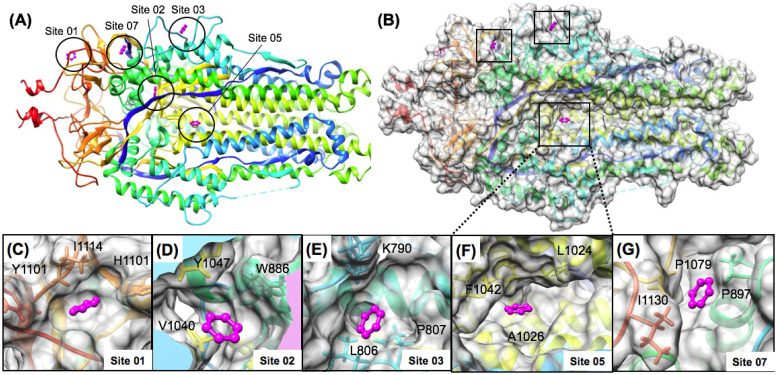
The 2019 emergence of, SARS-CoV-2 has tragically taken an immense toll on human life and far reaching impacts on society. There is a need to identify effective antivirals with diverse mechanisms of action in order to accelerate preclinical development. This study focused on five of the most established drug target proteins for direct acting small molecule antivirals: Nsp5 Main Protease, Nsp12 RNA-dependent RNA polymerase, Nsp13 Helicase, Nsp16 2'-O methyltransferase and the S2 subunit of the Spike protein. A workflow of solvent mapping and free energy calculations was used to identify and characterize favorable small-molecule binding sites for an aromatic pharmacophore (benzene). After identifying the most favorable sites, calculated ligand efficiencies were compared utilizing computational fragment screening. The most favorable sites overall were located on Nsp12 and Nsp16, whereas the most favorable sites for Nsp13 and S2 Spike had comparatively lower ligand efficiencies relative to Nsp12 and Nsp16. Utilizing fragment screening on numerous possible sites on Nsp13 helicase, we identified a favorable allosteric site on the N-terminal zinc binding domain (ZBD) that may be amenable to virtual or biophysical fragment screening efforts. Recent structural studies of the Nsp12:Nsp13 replication-transcription complex experimentally corroborates ligand binding at this site, which is revealed to be a functional Nsp8:Nsp13 protein-protein interaction site in the complex. Detailed structural analysis of Nsp13 ZBD conformations show the role of induced-fit flexibility in this ligand binding site and identify which conformational states are associated with efficient ligand binding. We hope that this map of over 200 possible small-molecule binding sites for these drug targets may be of use for ongoing discovery, design, and drug repurposing efforts. This information may be used to prioritize screening efforts or aid in the process of deciphering how a screening hit may bind to a specific target protein.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
