

SPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females
- PMID: 33596411
- PMCID: PMC8008487
- DOI: 10.1016/j.ajhg.2021.01.015
SPEN haploinsufficiency causes a neurodevelopmental disorder overlapping proximal 1p36 deletion syndrome with an episignature of X chromosomes in females
Abstract
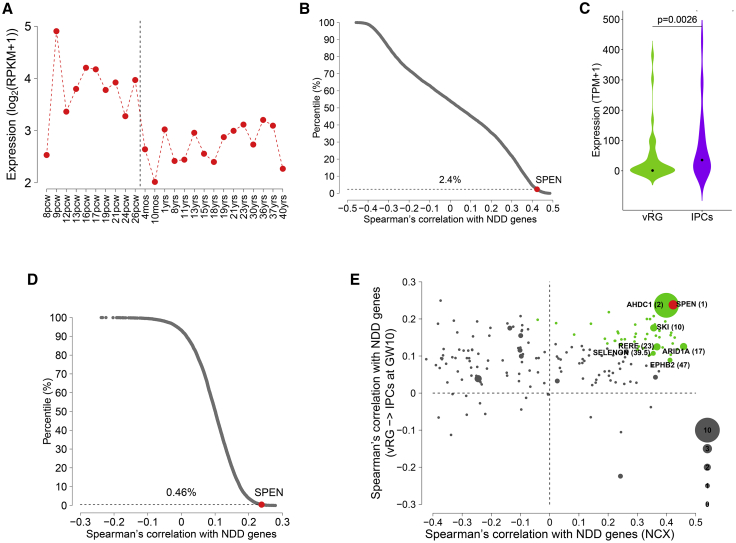
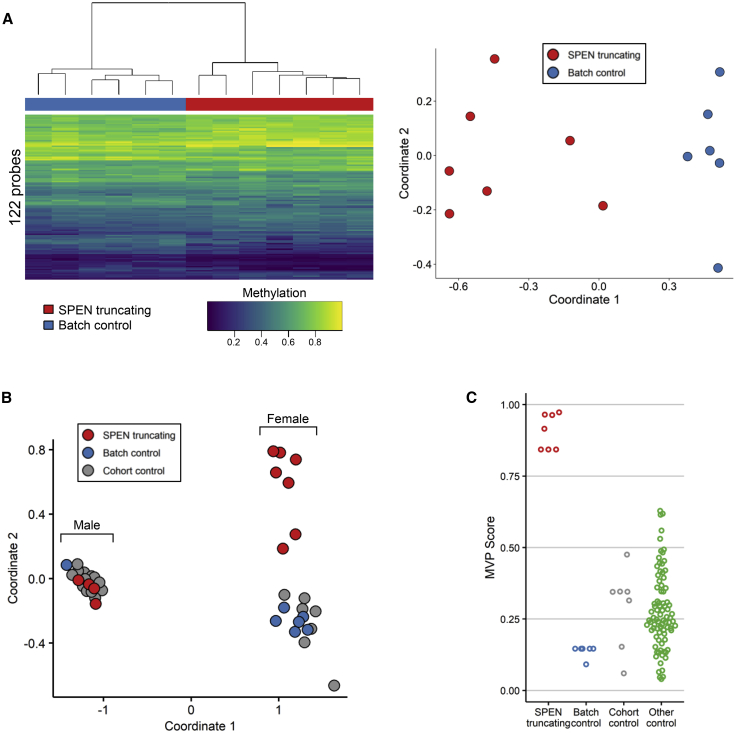
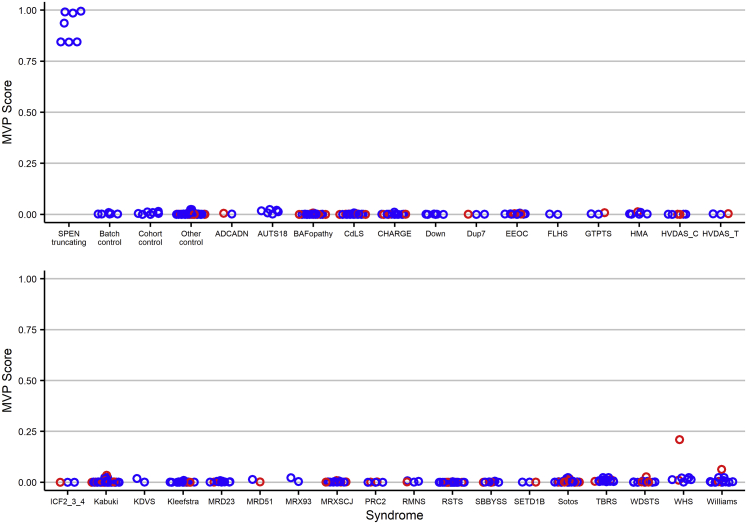
Deletion 1p36 (del1p36) syndrome is the most common human disorder resulting from a terminal autosomal deletion. This condition is molecularly and clinically heterogeneous. Deletions involving two non-overlapping regions, known as the distal (telomeric) and proximal (centromeric) critical regions, are sufficient to cause the majority of the recurrent clinical features, although with different facial features and dysmorphisms. SPEN encodes a transcriptional repressor commonly deleted in proximal del1p36 syndrome and is located centromeric to the proximal 1p36 critical region. Here, we used clinical data from 34 individuals with truncating variants in SPEN to define a neurodevelopmental disorder presenting with features that overlap considerably with those of proximal del1p36 syndrome. The clinical profile of this disease includes developmental delay/intellectual disability, autism spectrum disorder, anxiety, aggressive behavior, attention deficit disorder, hypotonia, brain and spine anomalies, congenital heart defects, high/narrow palate, facial dysmorphisms, and obesity/increased BMI, especially in females. SPEN also emerges as a relevant gene for del1p36 syndrome by co-expression analyses. Finally, we show that haploinsufficiency of SPEN is associated with a distinctive DNA methylation episignature of the X chromosome in affected females, providing further evidence of a specific contribution of the protein to the epigenetic control of this chromosome, and a paradigm of an X chromosome-specific episignature that classifies syndromic traits. We conclude that SPEN is required for multiple developmental processes and SPEN haploinsufficiency is a major contributor to a disorder associated with deletions centromeric to the previously established 1p36 critical regions.
Keywords: 1p36; DNA methylome analysis; SPEN; X chromosome; distal 1p36 deletion syndrome; episignature; genotype-phenotype correlations; neurodevelopmental disorder; obesity; proximal 1p36 deletion syndrome.
Copyright © 2021 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
M.D., K.Mc., K.G.M., and A.T. are employees of GeneDx. All the other authors declare no competing interests.
Figures

References
-
- Vissers L.E., de Ligt J., Gilissen C., Janssen I., Steehouwer M., de Vries P., van Lier B., Arts P., Wieskamp N., del Rosario M. A de novo paradigm for mental retardation. Nat. Genet. 2010;42:1109–1112. - PubMed
-
- Maulik P.K., Mascarenhas M.N., Mathers C.D., Dua T., Saxena S. Prevalence of intellectual disability: a meta-analysis of population-based studies. Res. Dev. Disabil. 2011;32:419–436. - PubMed
-
- Van Naarden Braun K., Christensen D., Doernberg N., Schieve L., Rice C., Wiggins L., Schendel D., Yeargin-Allsopp M. Trends in the prevalence of autism spectrum disorder, cerebral palsy, hearing loss, intellectual disability, and vision impairment, metropolitan atlanta, 1991-2010. PLoS ONE. 2015;10:e0124120. - PMC - PubMed
-
- Vissers L.E., Gilissen C., Veltman J.A. Genetic studies in intellectual disability and related disorders. Nat. Rev. Genet. 2016;17:9–18. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
