

Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models
- PMID: 33597034
- PMCID: PMC7888189
- DOI: 10.1186/s13321-020-00479-8
Could graph neural networks learn better molecular representation for drug discovery? A comparison study of descriptor-based and graph-based models
Abstract
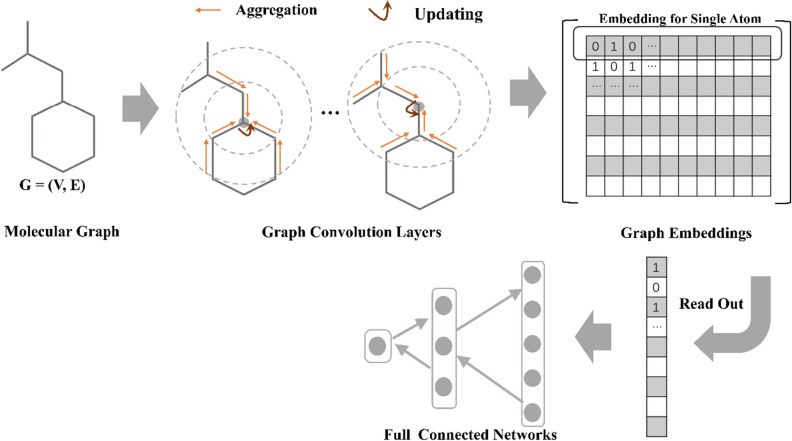
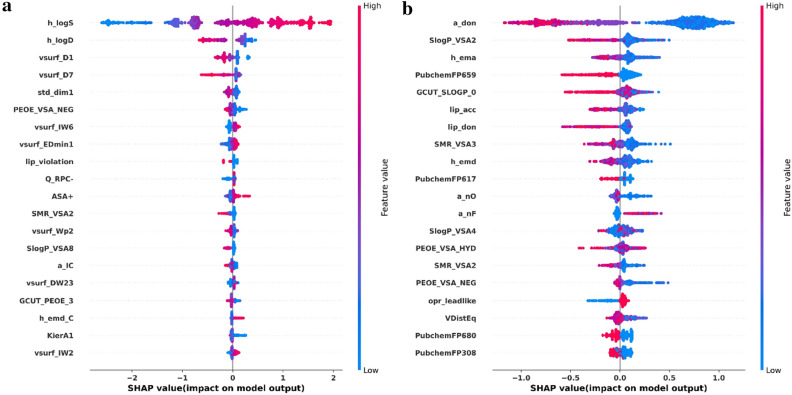
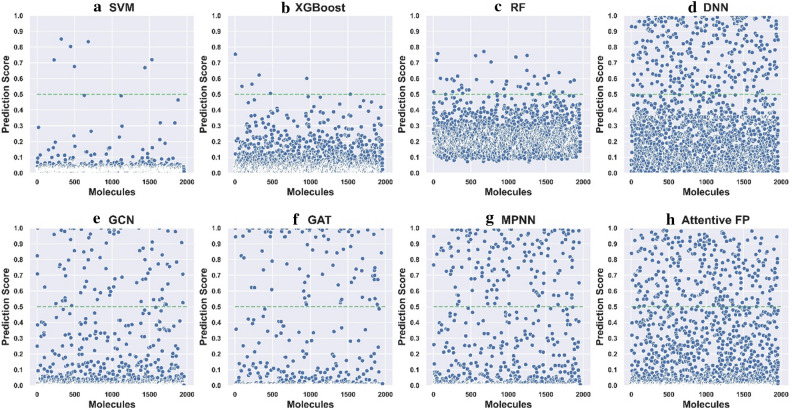
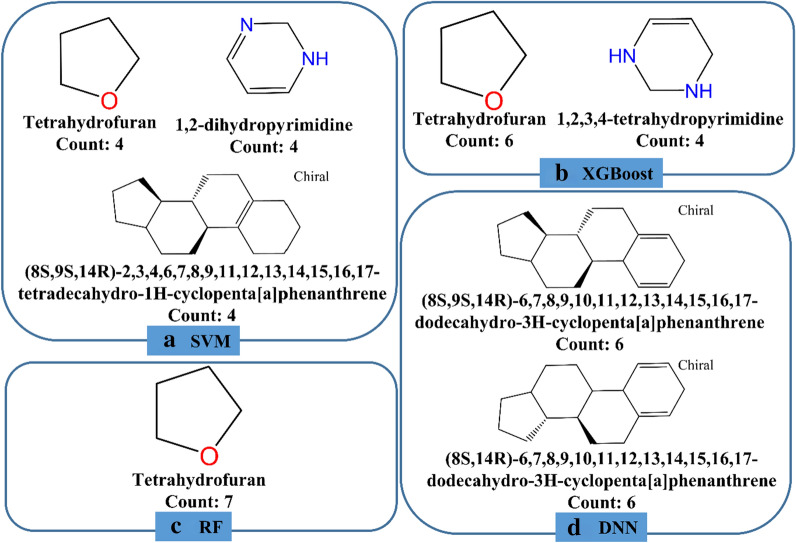
Graph neural networks (GNN) has been considered as an attractive modelling method for molecular property prediction, and numerous studies have shown that GNN could yield more promising results than traditional descriptor-based methods. In this study, based on 11 public datasets covering various property endpoints, the predictive capacity and computational efficiency of the prediction models developed by eight machine learning (ML) algorithms, including four descriptor-based models (SVM, XGBoost, RF and DNN) and four graph-based models (GCN, GAT, MPNN and Attentive FP), were extensively tested and compared. The results demonstrate that on average the descriptor-based models outperform the graph-based models in terms of prediction accuracy and computational efficiency. SVM generally achieves the best predictions for the regression tasks. Both RF and XGBoost can achieve reliable predictions for the classification tasks, and some of the graph-based models, such as Attentive FP and GCN, can yield outstanding performance for a fraction of larger or multi-task datasets. In terms of computational cost, XGBoost and RF are the two most efficient algorithms and only need a few seconds to train a model even for a large dataset. The model interpretations by the SHAP method can effectively explore the established domain knowledge for the descriptor-based models. Finally, we explored use of these models for virtual screening (VS) towards HIV and demonstrated that different ML algorithms offer diverse VS profiles. All in all, we believe that the off-the-shelf descriptor-based models still can be directly employed to accurately predict various chemical endpoints with excellent computability and interpretability.
Keywords: ADME/T prediction; Deep learning; Ensemble learning; Extreme gradient boosting; Graph neural networks.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Hou T, Li Y, Zhang W, et al. Recent developments of in silico predictions of intestinal absorption and oral bioavailability. Comb Chem High Throughput Screening. 2009;12:497–506. - PubMed
-
- Xia XY, Maliski EG, Gallant P, et al. Classification of kinase inhibitors using a Bayesian model. J Med Chem. 2004;47:4463–4470. - PubMed
-
- Tian S, Wang J, Li Y, et al. Drug-likeness analysis of traditional chinese medicines: prediction of drug-likeness using machine learning approaches. Mol Pharm. 2012;9:2875–2886. - PubMed
-
- Li D, Chen L, Li Y, et al. ADMET Evaluation in Drug Discovery. 13. Development of in silico prediction models for P-Glycoprotein Substrates. Mol Pharm. 2014;11:716–726. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
