

APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function
- PMID: 33597265
- PMCID: PMC8128342
- DOI: 10.1126/scitranslmed.abd7522
APOE immunotherapy reduces cerebral amyloid angiopathy and amyloid plaques while improving cerebrovascular function
Abstract
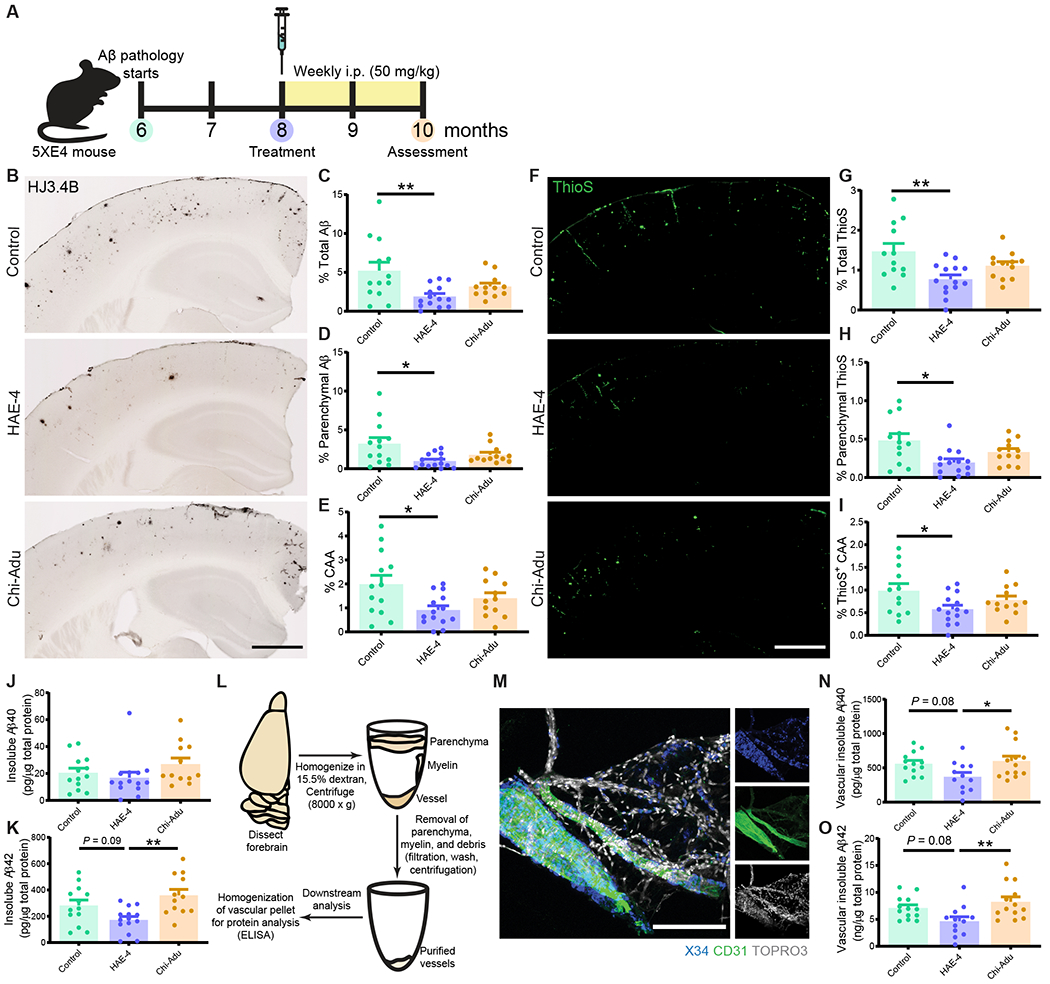
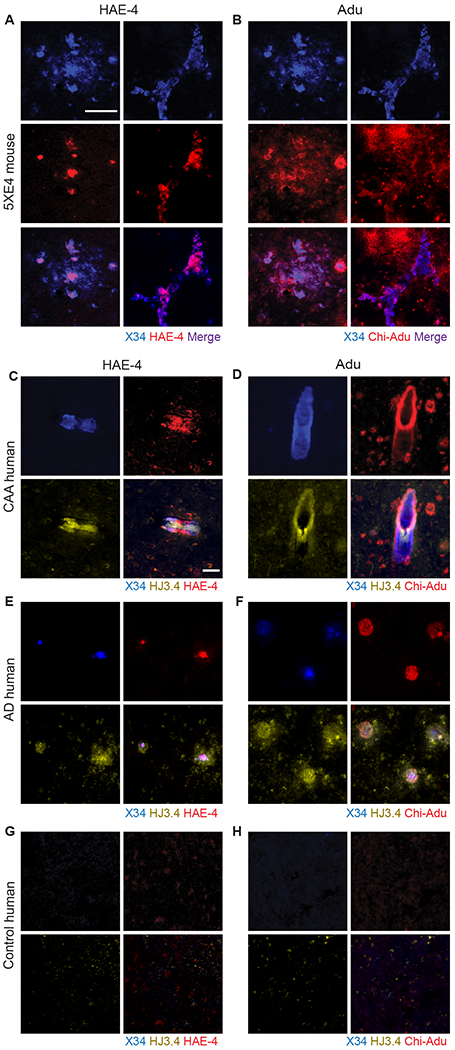
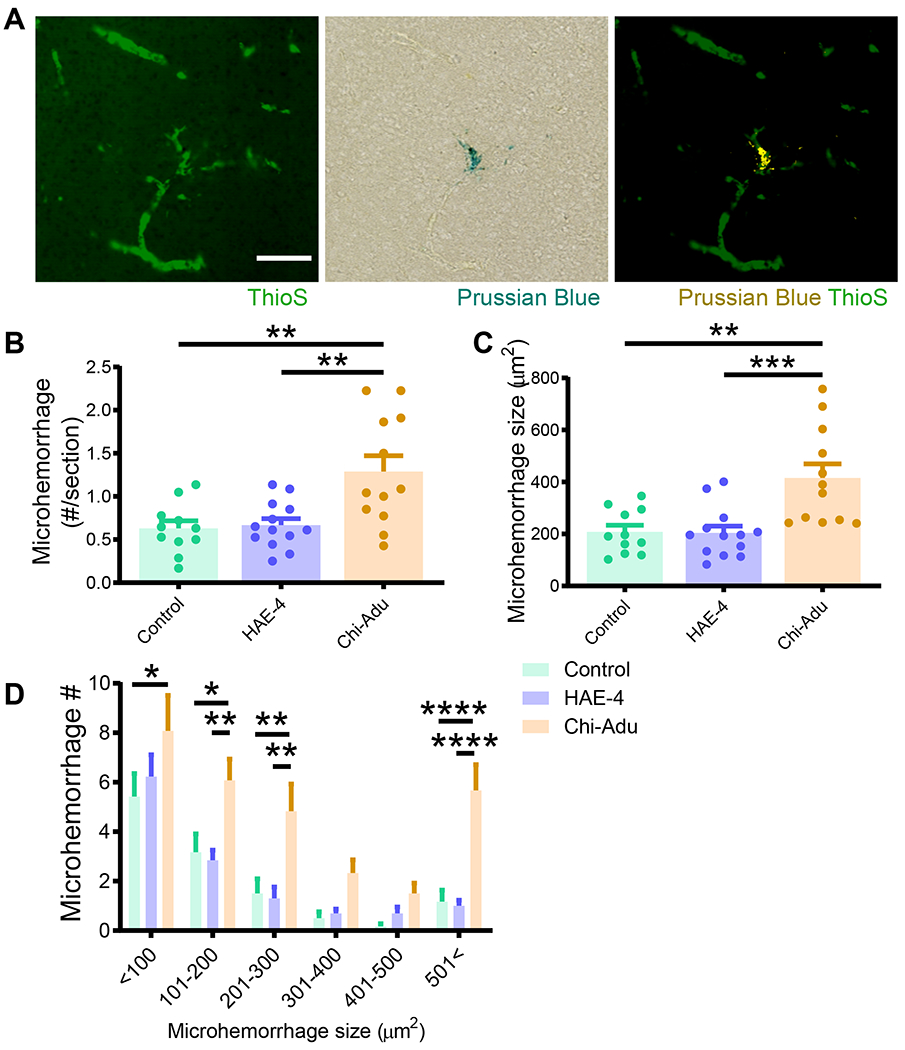
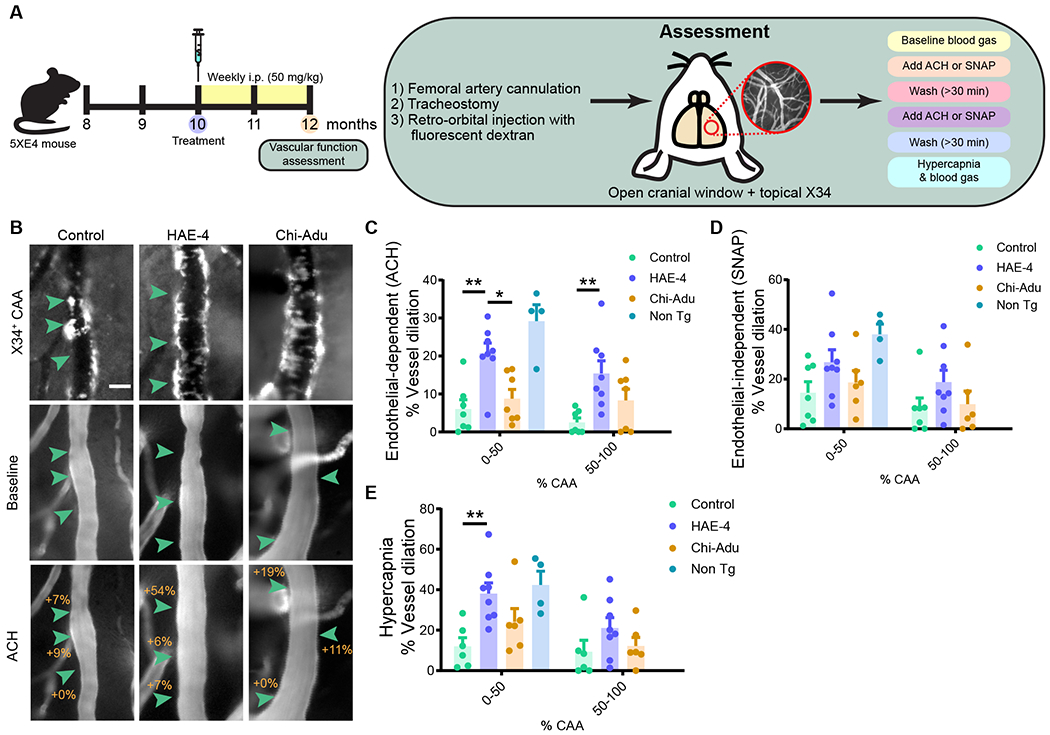
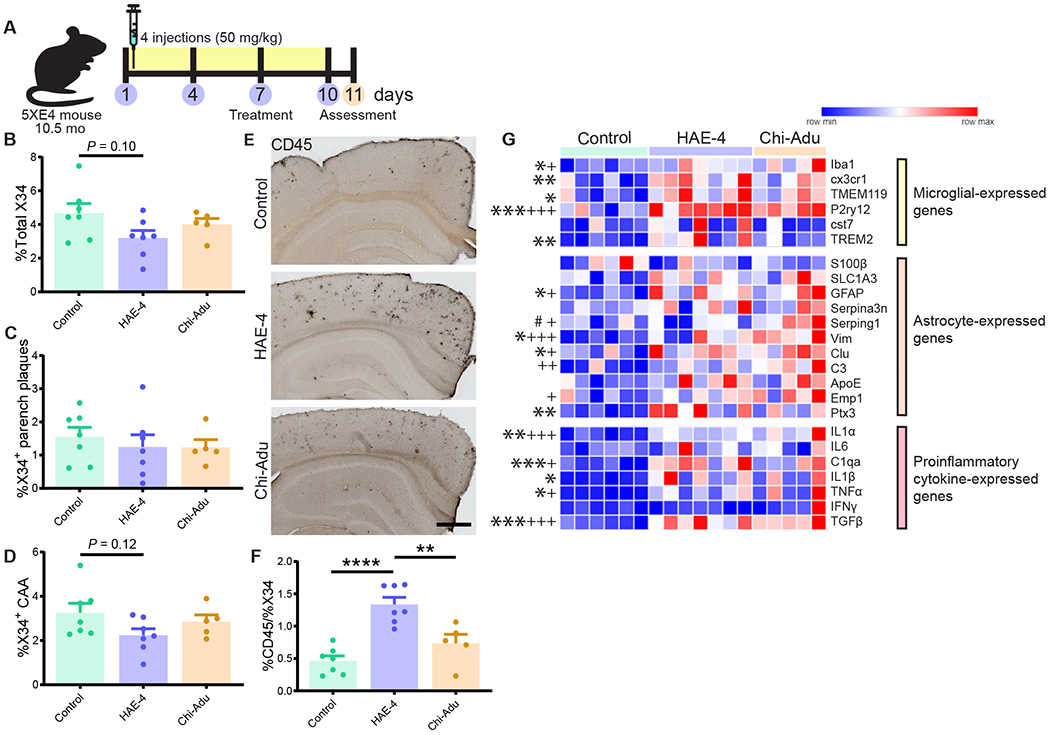
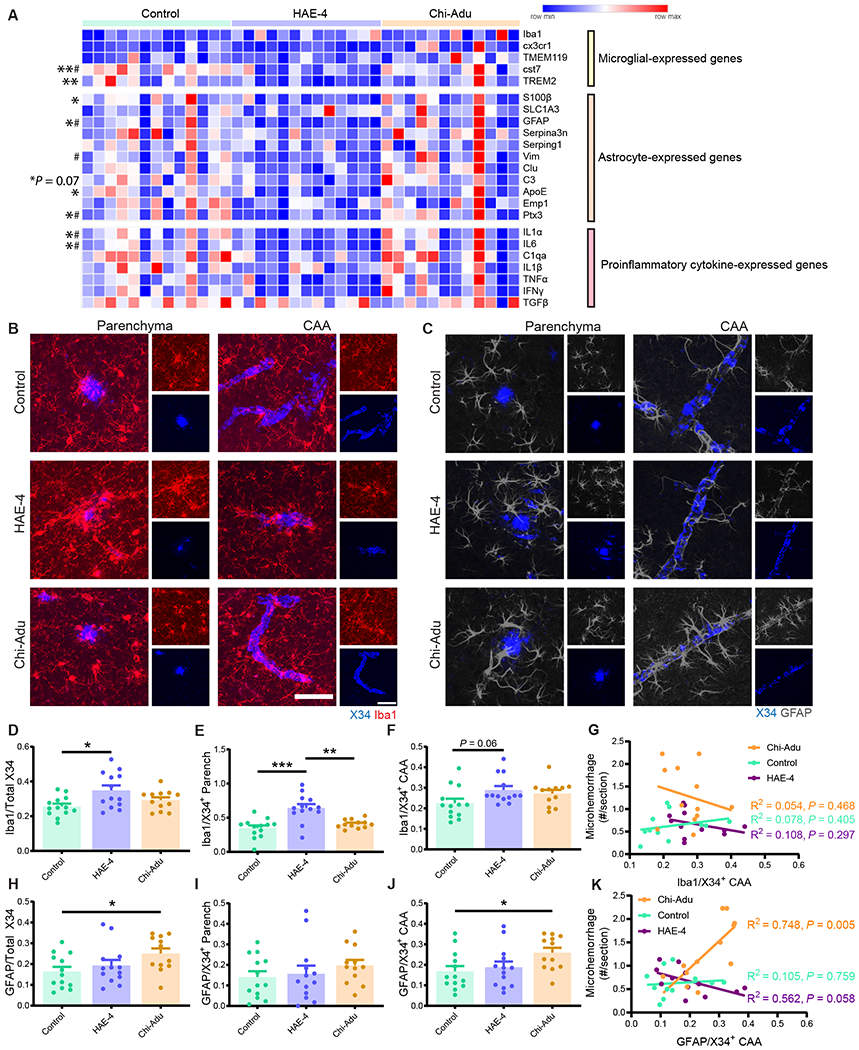
The ε4 allele of the apolipoprotein E (APOE) gene is the strongest genetic risk factor for late-onset Alzheimer's disease (AD) and greatly influences the development of amyloid-β (Aβ) pathology. Our current study investigated the potential therapeutic effects of the anti-human APOE antibody HAE-4, which selectively recognizes human APOE that is co-deposited with Aβ in cerebral amyloid angiopathy (CAA) and parenchymal amyloid pathology. In addition, we tested whether HAE-4 provoked brain hemorrhages, a component of amyloid-related imaging abnormalities (ARIA). ARIA is an adverse effect secondary to treatment with anti-Aβ antibodies that can occur in blood vessels with CAA. We used 5XFAD mice expressing human APOE4 +/+ (5XE4) that have prominent CAA and parenchymal plaque pathology to assess the efficacy of HAE-4 compared to an Aβ antibody that removes parenchymal Aβ but increases ARIA in humans. In chronically treated 5XE4 mice, HAE-4 reduced Aβ deposition including CAA compared to a control antibody, whereas the anti-Aβ antibody had no effect on CAA. Furthermore, the anti-Aβ antibody exacerbated microhemorrhage severity, which highly correlated with reactive astrocytes surrounding CAA. In contrast, HAE-4 did not stimulate microhemorrhages and instead rescued CAA-induced cerebrovascular dysfunction in leptomeningeal arteries in vivo. HAE-4 not only reduced amyloid but also dampened reactive microglial, astrocytic, and proinflammatory-associated genes in the cortex. These results suggest that targeting APOE in the core of both CAA and plaques could ameliorate amyloid pathology while protecting cerebrovascular integrity and function.
Copyright © 2021 The Authors, some rights reserved; exclusive licensee American Association for the Advancement of Science. No claim to original U.S. Government Works.
Conflict of interest statement
Competing interests
D.M.H. and H.J. are listed as inventors on US patent application #20190270794 entitled “Anti-APOE antibodies” from Washington University on APOE antibodies. N.B.L and R.J.W. are employees at Denali. A.P.S. is an employee of Codexis but conducted this work at Denali. D.M.H. co-founded and is on the scientific advisory board of C2N Diagnostics. D.M.H. is on the scientific advisory board of Denali and consults for Genentech, Merck, and Idorsia. Washington University (D.M.H.) has a sponsored research agreement to work on APOE antibodies from NextCure. All other authors have no competing interests.
Figures
Comment in
-
Antibody targets apolipoprotein E to remove amyloid-β deposits.Nat Rev Neurol. 2021 Apr;17(4):194. doi: 10.1038/s41582-021-00479-8. Nat Rev Neurol. 2021. PMID: 33750931 No abstract available.
References
-
- Sperling R, Salloway S, Brooks DJ, Tampieri D, Barakos J, Fox NC, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Lieberburg I, Arrighi HM, Morris KA, Lu Y, Liu E, Gregg KM, Brashear HR, Kinney GG, Black R, Grundman M, Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: A retrospective analysis, Lancet Neurol. 11, 241–249 (2012). - PMC - PubMed
-
- Sperling RA, Jack CR, Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, Black RS, Brashear HR, Grundman M, Siemers ER, Feldman HH, Schindler RJ, Amyloid-related imaging abnormalities in amyloid-modifying therapeutic trials: Recommendations from the Alzheimer’s Association Research Roundtable Workgroup, Alzheimer’s Dement. 7, 367–385 (2011). - PMC - PubMed
-
- Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D, Passive immunotherapy against Aβ in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage, J. Neuroinflammation 1, 1–11 (2004). - PMC - PubMed
-
- Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, Holtzman DM, Bales KR, Gitter BD, May PC, Paul SM, DeMattos RB, Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid β, J. Neurosci. 25, 629–636 (2005). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
