

SOX9 keeps growth plates and articular cartilage healthy by inhibiting chondrocyte dedifferentiation/osteoblastic redifferentiation
- PMID: 33597301
- PMCID: PMC7923381
- DOI: 10.1073/pnas.2019152118
SOX9 keeps growth plates and articular cartilage healthy by inhibiting chondrocyte dedifferentiation/osteoblastic redifferentiation
Abstract
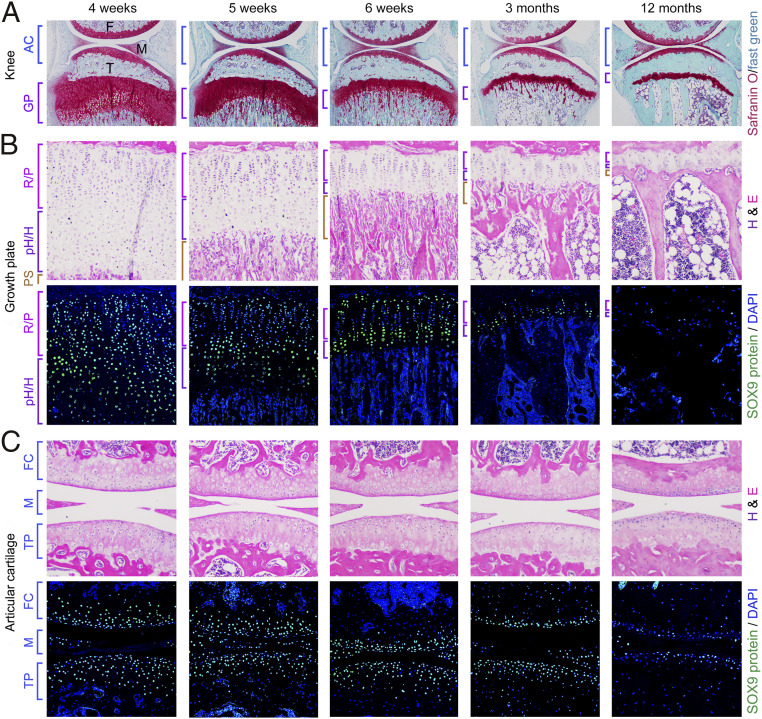
Cartilage is essential throughout vertebrate life. It starts developing in embryos when osteochondroprogenitor cells commit to chondrogenesis, activate a pancartilaginous program to form cartilaginous skeletal primordia, and also embrace a growth-plate program to drive skeletal growth or an articular program to build permanent joint cartilage. Various forms of cartilage malformation and degeneration diseases afflict humans, but underlying mechanisms are still incompletely understood and treatment options suboptimal. The transcription factor SOX9 is required for embryonic chondrogenesis, but its postnatal roles remain unclear, despite evidence that it is down-regulated in osteoarthritis and heterozygously inactivated in campomelic dysplasia, a severe skeletal dysplasia characterized postnatally by small stature and kyphoscoliosis. Using conditional knockout mice and high-throughput sequencing assays, we show here that SOX9 is required postnatally to prevent growth-plate closure and preosteoarthritic deterioration of articular cartilage. Its deficiency prompts growth-plate chondrocytes at all stages to swiftly reach a terminal/dedifferentiated stage marked by expression of chondrocyte-specific (Mgp) and progenitor-specific (Nt5e and Sox4) genes. Up-regulation of osteogenic genes (Runx2, Sp7, and Postn) and overt osteoblastogenesis quickly ensue. SOX9 deficiency does not perturb the articular program, except in load-bearing regions, where it also provokes chondrocyte-to-osteoblast conversion via a progenitor stage. Pathway analyses support roles for SOX9 in controlling TGFβ and BMP signaling activities during this cell lineage transition. Altogether, these findings deepen our current understanding of the cellular and molecular mechanisms that specifically ensure lifelong growth-plate and articular cartilage vigor by identifying osteogenic plasticity of growth-plate and articular chondrocytes and a SOX9-countered chondrocyte dedifferentiation/osteoblast redifferentiation process.
Keywords: SOX9; cartilage; cell differentiation; lineage determination; transcriptional regulation.
Conflict of interest statement
The authors declare no competing interest.
Figures







Comment in
-
Keep your Sox on, chondrocytes!Nat Rev Rheumatol. 2021 Jul;17(7):383-384. doi: 10.1038/s41584-021-00628-9. Nat Rev Rheumatol. 2021. PMID: 33953371 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
