

Expanded catalog of microbial genes and metagenome-assembled genomes from the pig gut microbiome
- PMID: 33597514
- PMCID: PMC7889623
- DOI: 10.1038/s41467-021-21295-0
Expanded catalog of microbial genes and metagenome-assembled genomes from the pig gut microbiome
Abstract
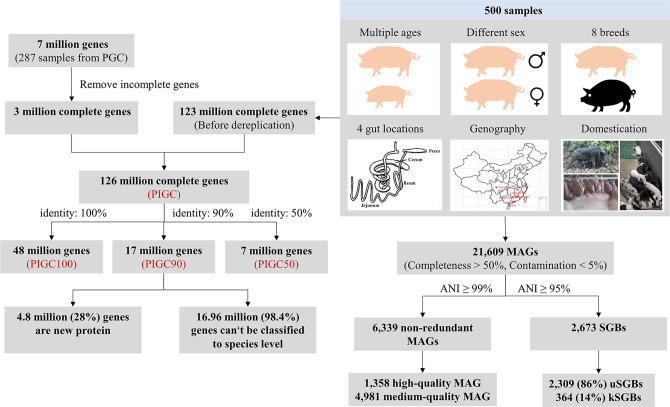
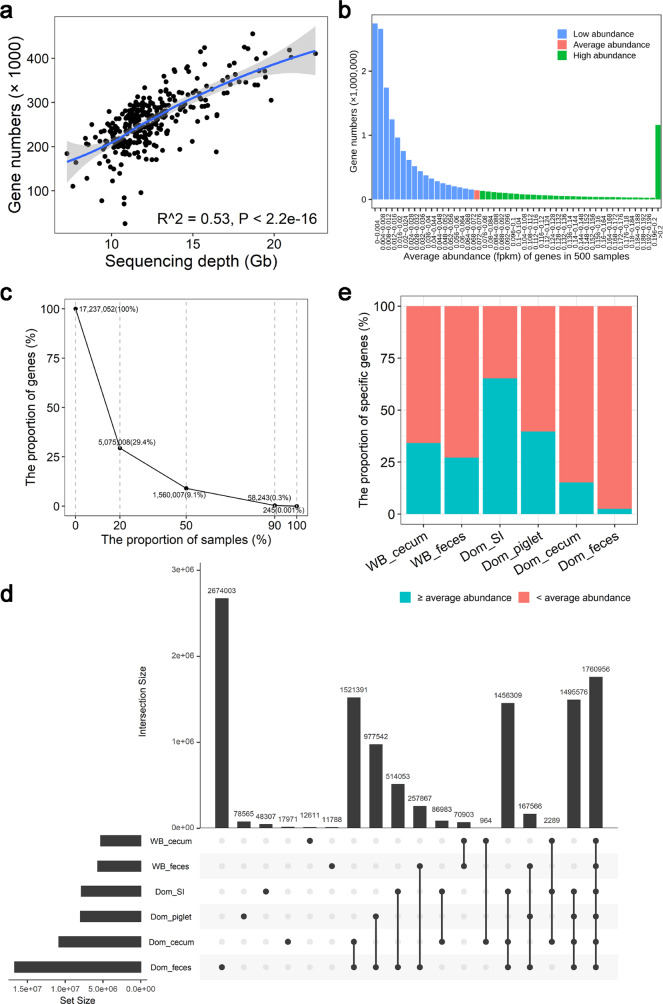
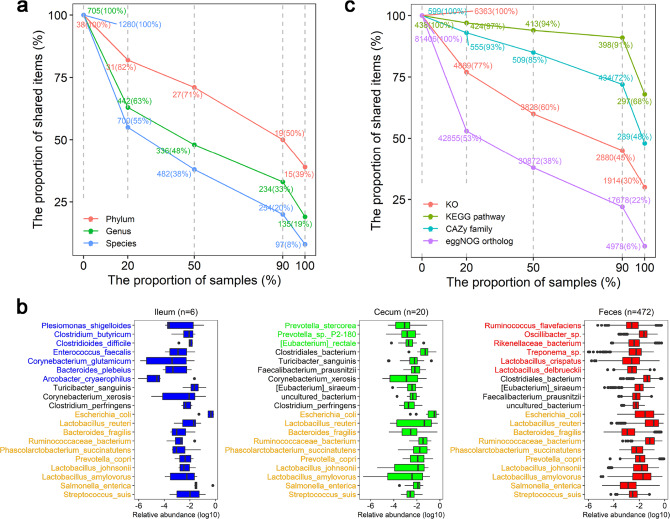
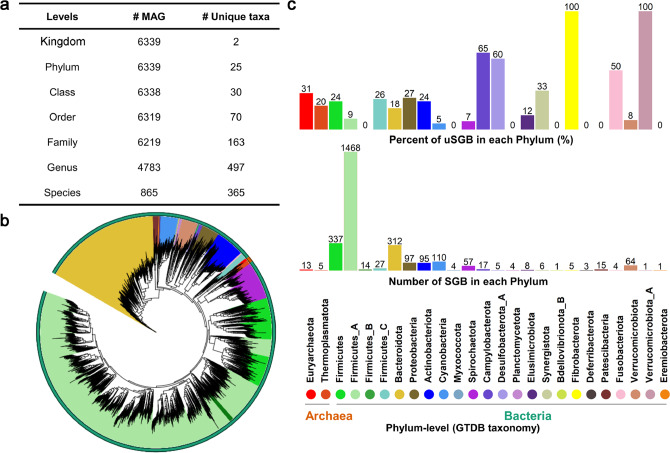
Gut microbiota plays an important role in pig health and production. Still, availability of sequenced genomes and functional information for most pig gut microbes remains limited. Here we perform a landscape survey of the swine gut microbiome, spanning extensive sample sources by deep metagenomic sequencing resulting in an expanded gene catalog named pig integrated gene catalog (PIGC), containing 17,237,052 complete genes clustered at 90% protein identity from 787 gut metagenomes, of which 28% are unknown proteins. Using binning analysis, 6339 metagenome-assembled genomes (MAGs) were obtained, which were clustered to 2673 species-level genome bins (SGBs), among which 86% (2309) SGBs are unknown based on current databases. Using the present gene catalog and MAGs, we identified several strain-level differences between the gut microbiome of wild boars and commercial Duroc pigs. PIGC and MAGs provide expanded resources for swine gut microbiome-related research.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
