

Determinants of genome-wide distribution and evolution of uORFs in eukaryotes
- PMID: 33597535
- PMCID: PMC7889888
- DOI: 10.1038/s41467-021-21394-y
Determinants of genome-wide distribution and evolution of uORFs in eukaryotes
Erratum in
-
Author Correction: Determinants of genome-wide distribution and evolution of uORFs in eukaryotes.Nat Commun. 2021 Mar 31;12(1):2101. doi: 10.1038/s41467-021-22435-2. Nat Commun. 2021. PMID: 33790278 Free PMC article. No abstract available.
Abstract
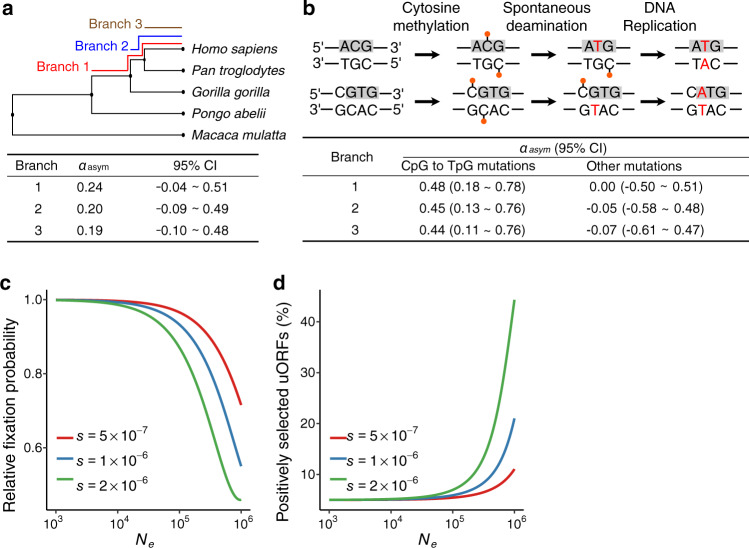
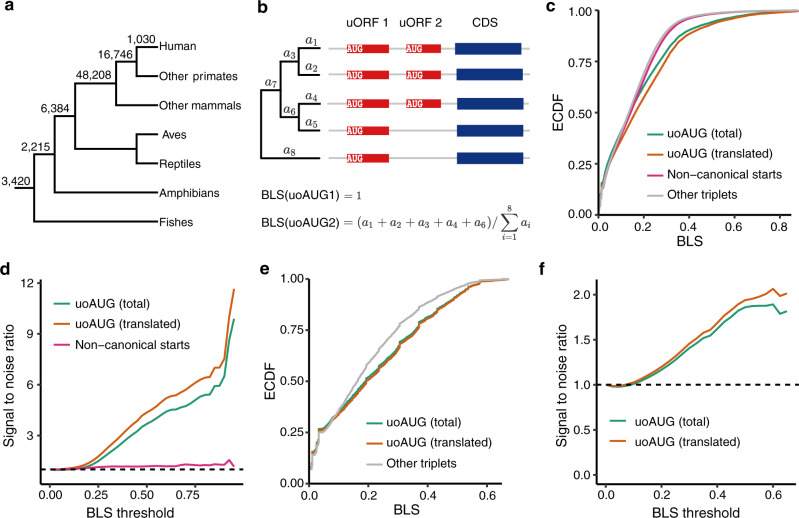
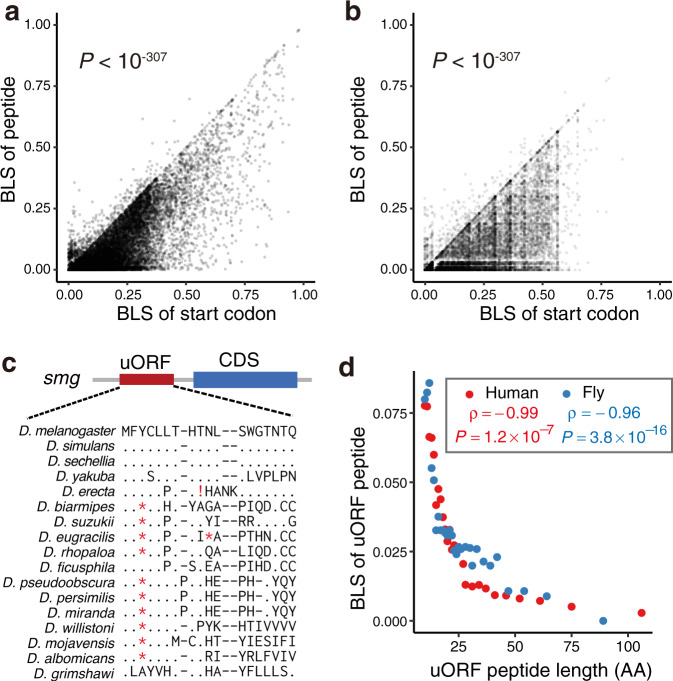
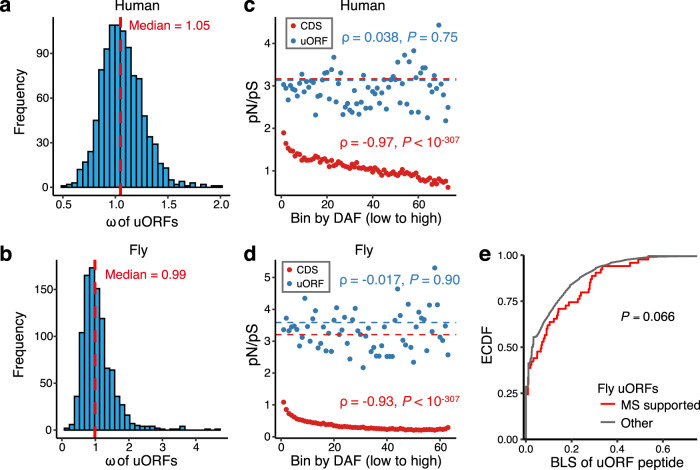
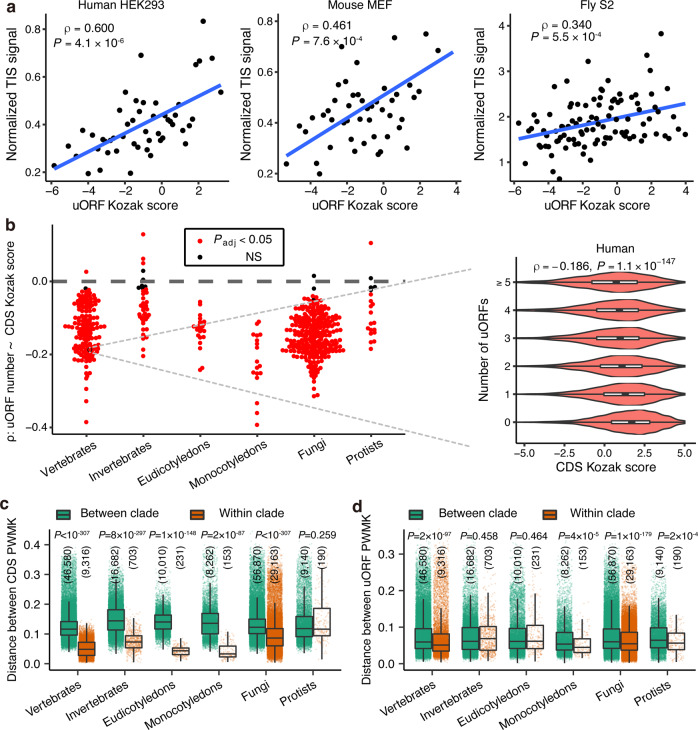
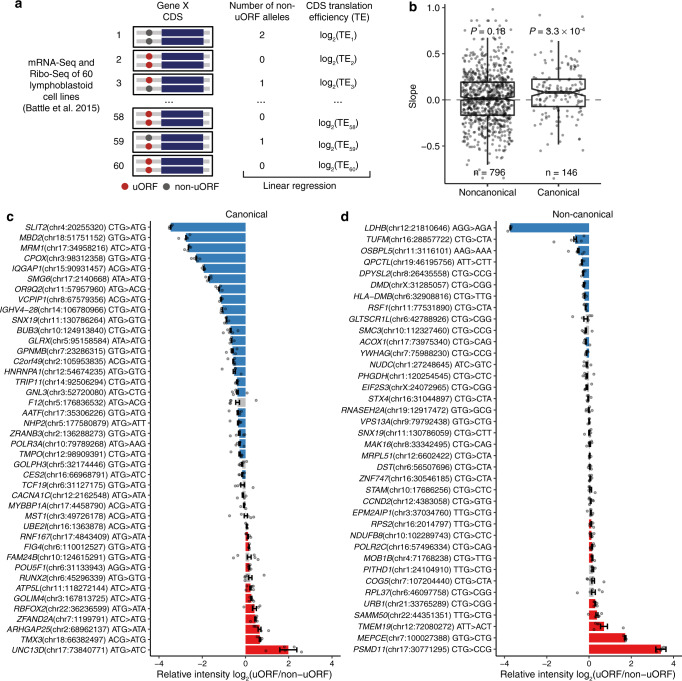
Upstream open reading frames (uORFs) play widespread regulatory functions in modulating mRNA translation in eukaryotes, but the principles underlying the genomic distribution and evolution of uORFs remain poorly understood. Here, we analyze ~17 million putative canonical uORFs in 478 eukaryotic species that span most of the extant taxa of eukaryotes. We demonstrate how positive and purifying selection, coupled with differences in effective population size (Ne), has shaped the contents of uORFs in eukaryotes. Besides, gene expression level is important in influencing uORF occurrences across genes in a species. Our analyses suggest that most uORFs might play regulatory roles rather than encode functional peptides. We also show that the Kozak sequence context of uORFs has evolved across eukaryotic clades, and that noncanonical uORFs tend to have weaker suppressive effects than canonical uORFs in translation regulation. This study provides insights into the driving forces underlying uORF evolution in eukaryotes.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
