

Kallmann Syndrome Due to Heterozygous Mutation in SOX10 Coexisting With Waardenburg Syndrome Type II: Case Report and Review of Literature
- PMID: 33597923
- PMCID: PMC7883637
- DOI: 10.3389/fendo.2020.592831
Kallmann Syndrome Due to Heterozygous Mutation in SOX10 Coexisting With Waardenburg Syndrome Type II: Case Report and Review of Literature
Abstract
Introduction: Kallmann syndrome (KS) is idiopathic hypogonadotropic hypogonadism with olfactory loss or decline. Waardenburg syndrome type II (WS2) is a clinically and genetically heterogeneous disease, characterized by congenital sensorineural deafness and abnormal pigmentation of the iris, hair, and skin. Recently, mutations in the well-known WS pathogenic gene SOX10 have been found in some KS patients with deafness, but whether SOX10 is a co-pathogenic gene of KS and WS remains uncertain. Here, we report a rare case of KS and WS2 co-occurrence due to SOX10 mutations.
Methods: Detailed histories were collected through questionnaires and physical examination. Blood samples of the patient and his family members were collected after obtaining informed consents. Suspected mutations were amplified and verified by Sanger sequencing after the next generation sequencing of related genes. The raw sequence data were compared to the known gene sequence data in publicly available sequence data bases using Burrows-Wheeler Aligner software (BWA, 0.7.12-r1039).
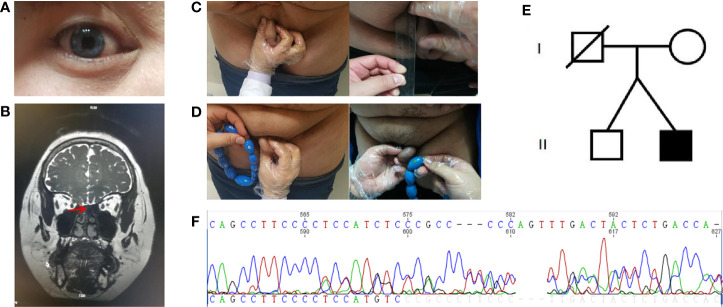
Results: A 28-year-old male patient sought treatment for hypogonadism and the absence of secondary sexual characteristics. In addition, he showed signs of obesity, hyposmia, sensorineural hearing loss, and blue iris. Magnetic resonance imaging (MRI) of the olfactory bulb showed small bilateral olfactory bulbs and tracts and diaphragma cerebri. MRI of the pituitary gland revealed a flat pituitary gland in the sella. Laboratory examination demonstrated hypogonadotropic hypogonadism, pituitary hypothyroidism, subclinical hypothyroidism, and the presence of insulin resistance with normal blood glucose levels. Sequencing of the SOX10 gene showed a 20 bp insertion in between coding bases 1,179 and 1,180 (c.1179_1180insACTATGGCTCAGCCTTCCCC). This results in a frame-shifting mutation of the 394th amino acid serine in exon4 with the resulting the amino acid sequence of the protein predicted to be TMAQPSP PSPAPSLTTL TISPQDPIMA TRARPLASTR PSPIWGPRSG PSTRPSLTPA PQGPSPTAPH TGSSQYIRHC PGPKGGPVAT TPRPAPAPSL CALFLAHLRP GGGSGGG*.
Conclusion: SOX10 plays an important role in some critical stages of neural crest cell development and SOX10 mutation may be a common pathogenic factor for both KS and WS. Therefore, SOX10 mutation analysis should be considered for KS patients with combined WS clinical manifestations, especially deafness.
Keywords: Kallmann syndrome; SOX10 mutations; Waardenburg syndrome type Ⅱ; co-occurrence; hypogonadotropic hypogonadism; sensorineural deafness.
Copyright © 2021 Chen, Wang and Lai.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Diagnosis and genetic analysis of a case of Waardenburg syndrome type 2 with hypogonadotropic hypogonadism caused by SOX10 gene deletion.Yi Chuan. 2022 Dec 20;44(12):1158-1166. doi: 10.16288/j.yczz.22-161. Yi Chuan. 2022. PMID: 36927561
-
De novo SOX10 Nonsense Mutation in a Patient with Kallmann Syndrome, Deafness, Iris Hypopigmentation, and Hyperthyroidism.Ann Clin Lab Sci. 2018 Mar;48(2):248-252. Ann Clin Lab Sci. 2018. PMID: 29678855
-
Loss-of-Function SOX10 Mutation in a Patient with Kallmann Syndrome, Hearing Loss, and Iris Hypopigmentation.Horm Res Paediatr. 2015;84(3):212-6. doi: 10.1159/000436965. Epub 2015 Jul 29. Horm Res Paediatr. 2015. PMID: 26228106
-
[Kallmann syndrome with deafness caused by SOX10 mutation: Advances in research].Zhonghua Nan Ke Xue. 2017 Sep;23(9):838-841. Zhonghua Nan Ke Xue. 2017. PMID: 29726667 Review. Chinese.
-
SOX10: 20 years of phenotypic plurality and current understanding of its developmental function.J Med Genet. 2022 Feb;59(2):105-114. doi: 10.1136/jmedgenet-2021-108105. Epub 2021 Oct 19. J Med Genet. 2022. PMID: 34667088 Free PMC article. Review.
Cited by
-
Molecular diagnosis of Kallmann syndrome with diabetes by whole exome sequencing and bioinformatic approaches.World J Diabetes. 2021 Dec 15;12(12):2058-2072. doi: 10.4239/wjd.v12.i12.2058. World J Diabetes. 2021. PMID: 35047120 Free PMC article.
-
Mutation spectrum of Kallmann syndrome: identification of five novel mutations across ANOS1 and FGFR1.Reprod Biol Endocrinol. 2023 Mar 1;21(1):23. doi: 10.1186/s12958-023-01074-w. Reprod Biol Endocrinol. 2023. PMID: 36859276 Free PMC article.
-
Sox10 is required for systemic initiation of bone mineralization.Development. 2025 Jan 15;152(2):dev204357. doi: 10.1242/dev.204357. Epub 2025 Jan 20. Development. 2025. PMID: 39791977 Free PMC article.
References
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources