

Regulation and metabolic functions of mTORC1 and mTORC2
- PMID: 33599151
- PMCID: PMC8424549
- DOI: 10.1152/physrev.00026.2020
Regulation and metabolic functions of mTORC1 and mTORC2
Abstract
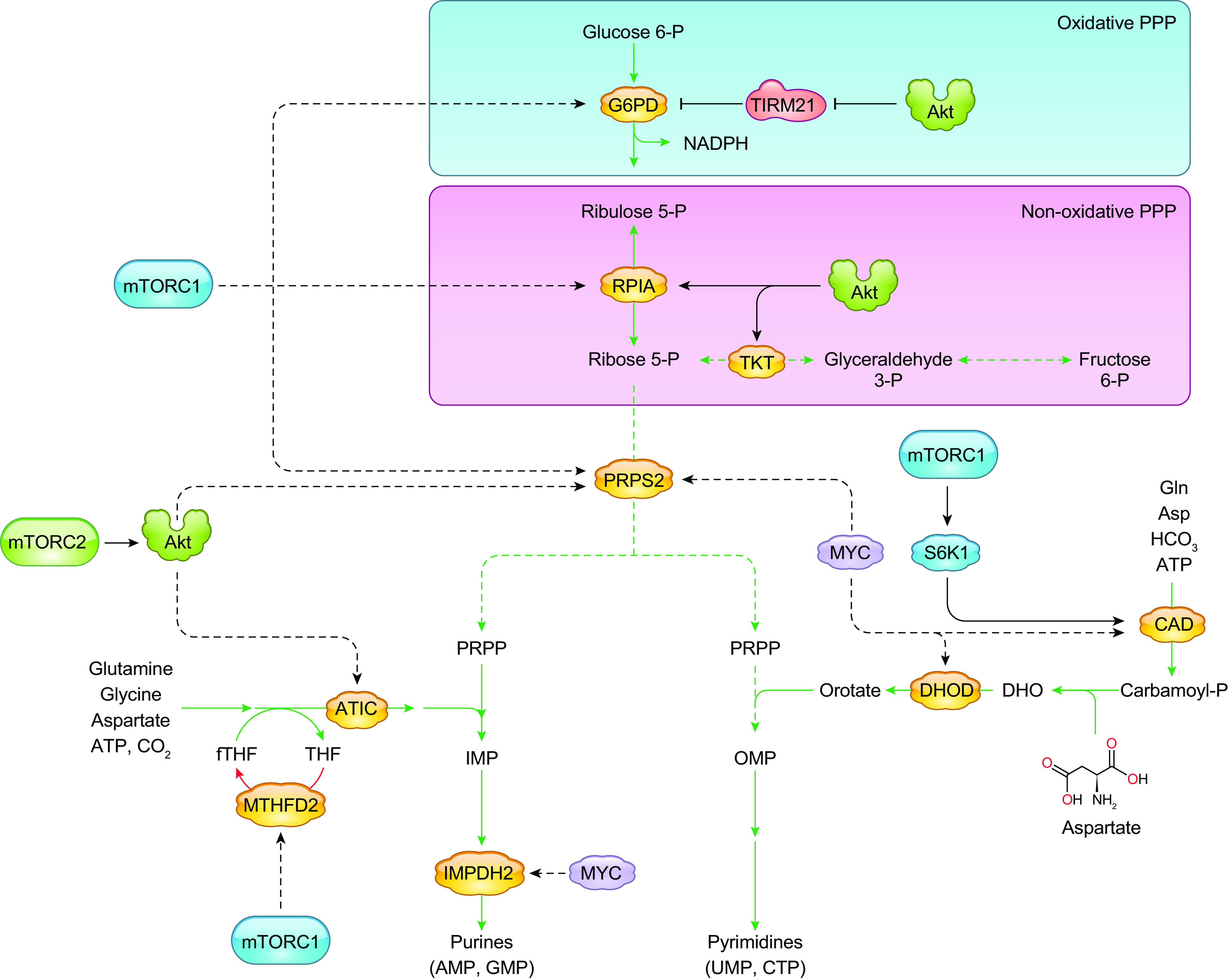
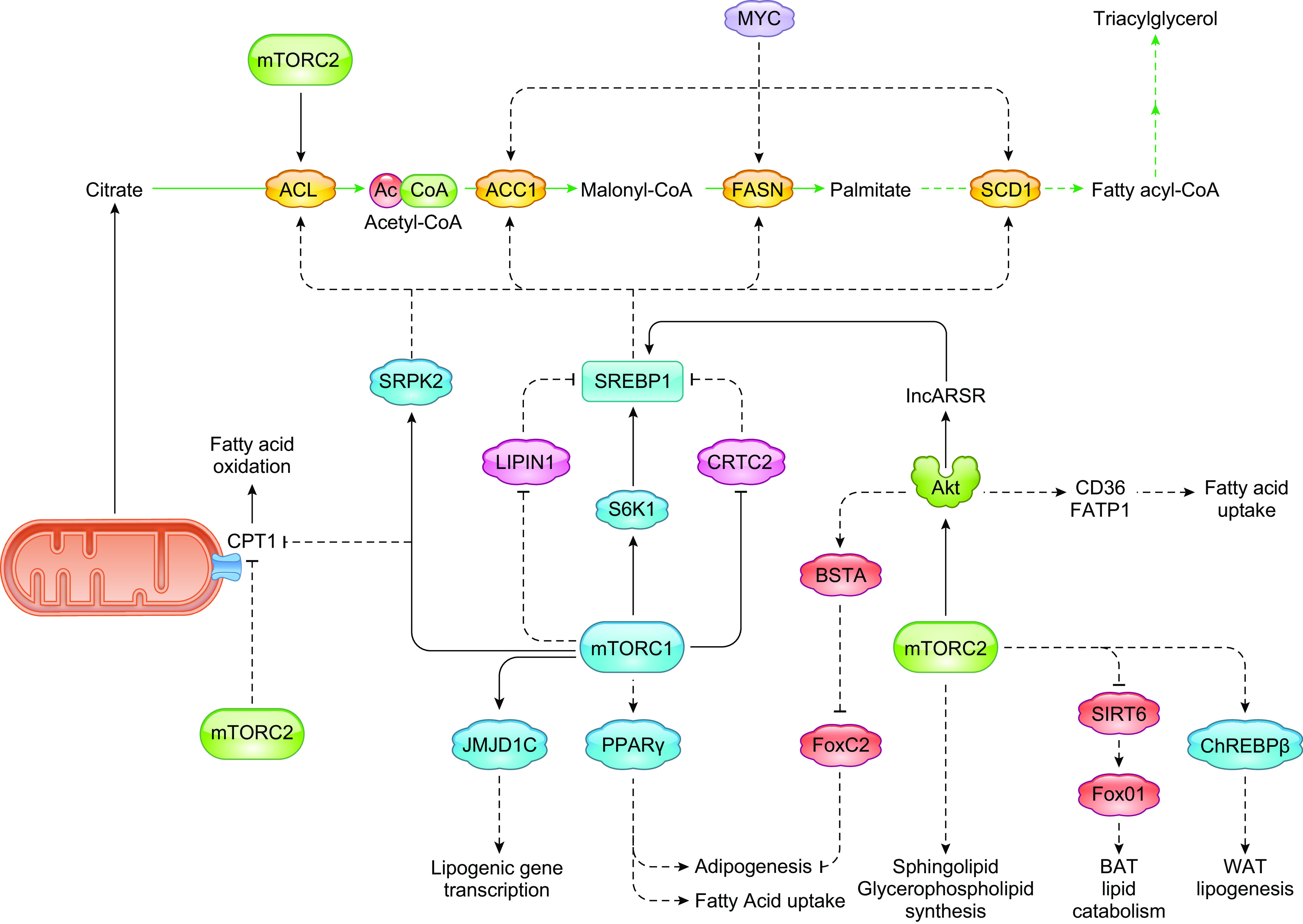
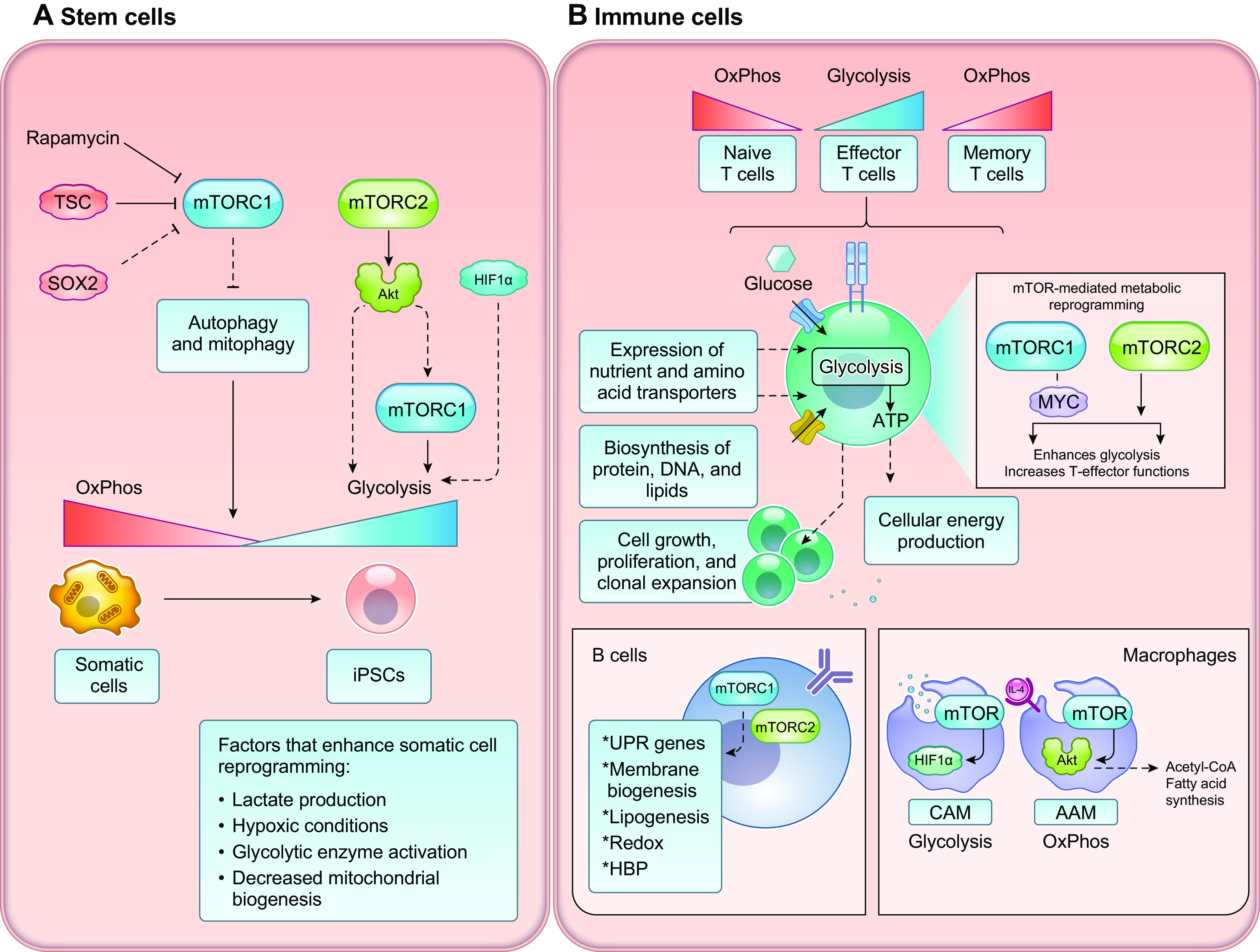
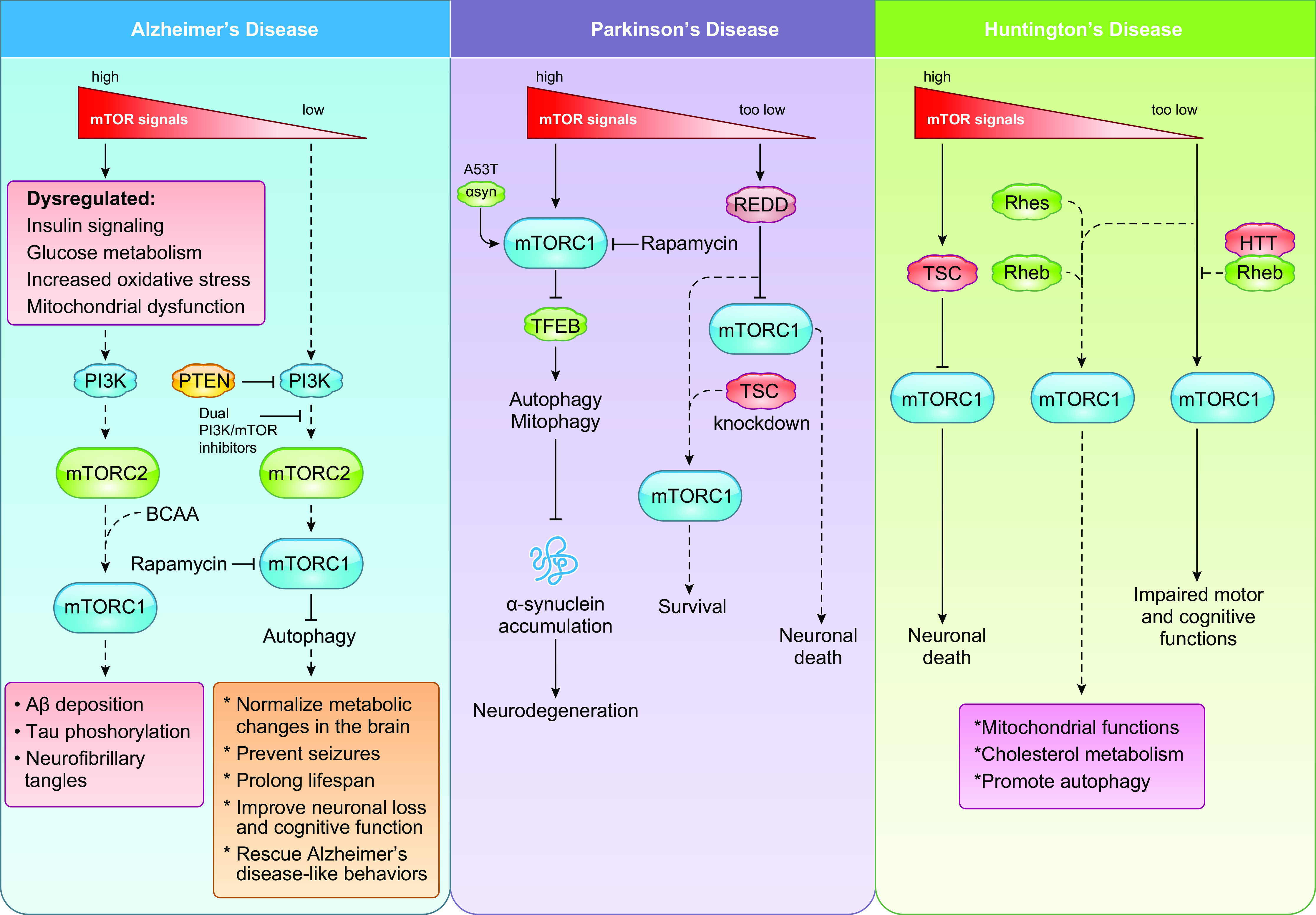
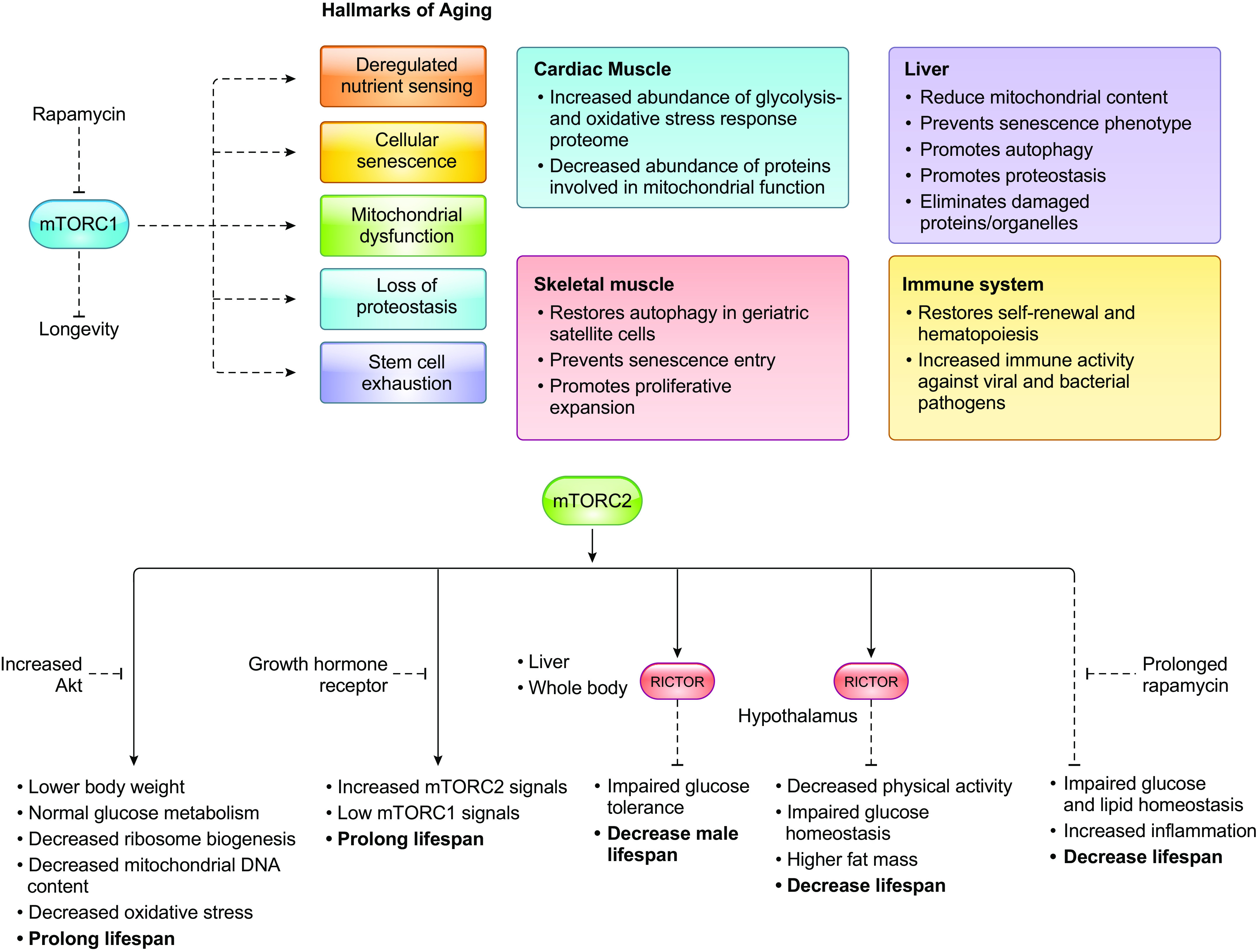
Cells metabolize nutrients for biosynthetic and bioenergetic needs to fuel growth and proliferation. The uptake of nutrients from the environment and their intracellular metabolism is a highly controlled process that involves cross talk between growth signaling and metabolic pathways. Despite constant fluctuations in nutrient availability and environmental signals, normal cells restore metabolic homeostasis to maintain cellular functions and prevent disease. A central signaling molecule that integrates growth with metabolism is the mechanistic target of rapamycin (mTOR). mTOR is a protein kinase that responds to levels of nutrients and growth signals. mTOR forms two protein complexes, mTORC1, which is sensitive to rapamycin, and mTORC2, which is not directly inhibited by this drug. Rapamycin has facilitated the discovery of the various functions of mTORC1 in metabolism. Genetic models that disrupt either mTORC1 or mTORC2 have expanded our knowledge of their cellular, tissue, as well as systemic functions in metabolism. Nevertheless, our knowledge of the regulation and functions of mTORC2, particularly in metabolism, has lagged behind. Since mTOR is an important target for cancer, aging, and other metabolism-related pathologies, understanding the distinct and overlapping regulation and functions of the two mTOR complexes is vital for the development of more effective therapeutic strategies. This review discusses the key discoveries and recent findings on the regulation and metabolic functions of the mTOR complexes. We highlight findings from cancer models but also discuss other examples of the mTOR-mediated metabolic reprogramming occurring in stem and immune cells, type 2 diabetes/obesity, neurodegenerative disorders, and aging.
Keywords: cancer metabolism; glycolysis; lipid metabolism; mTOR; metabolic reprogramming.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
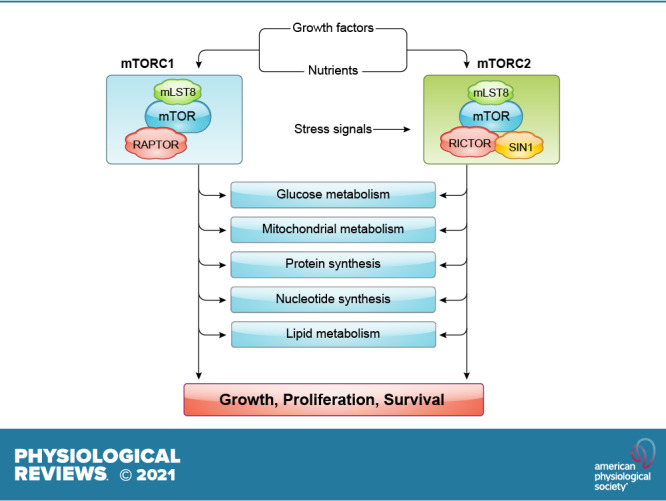
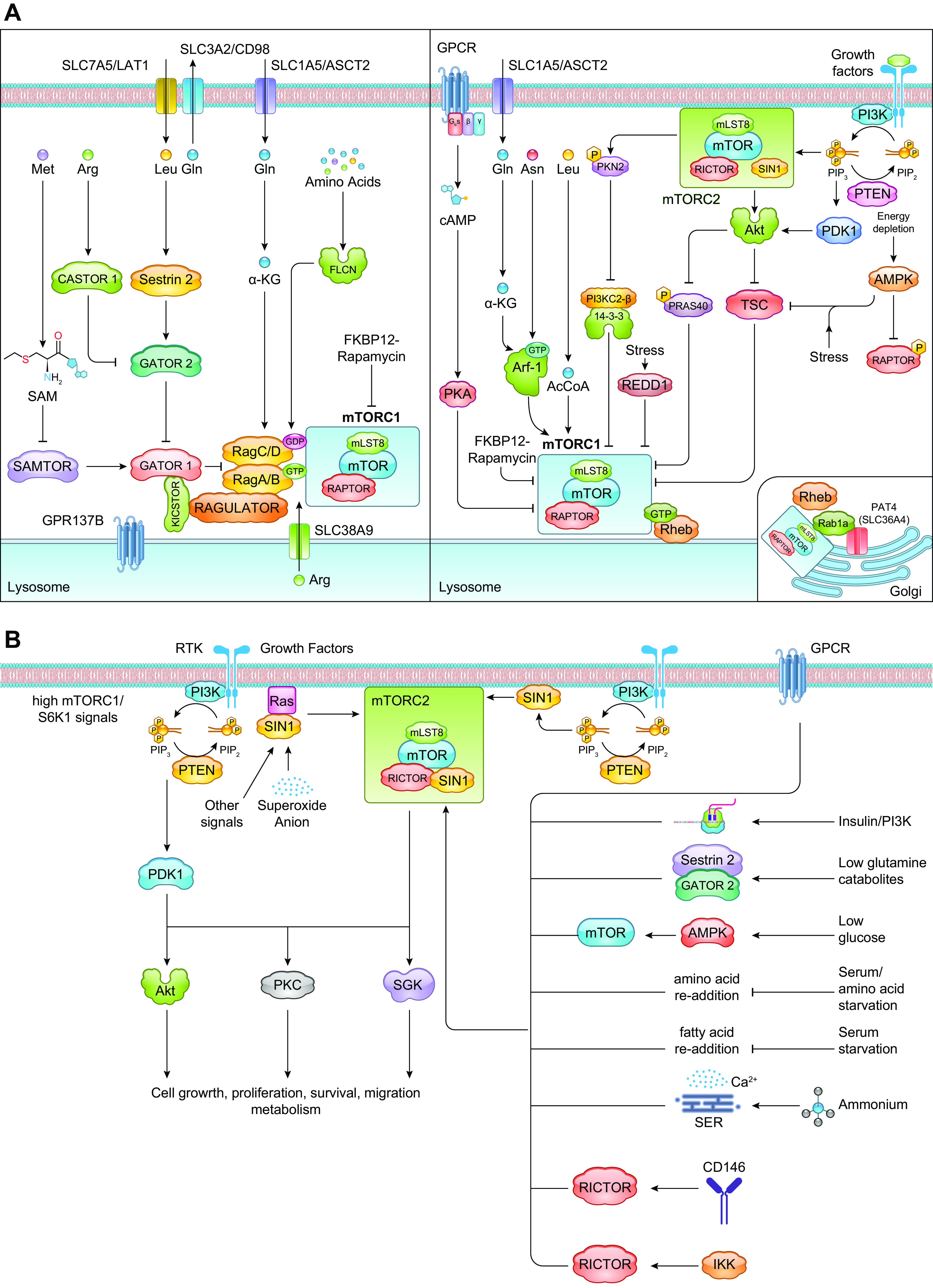
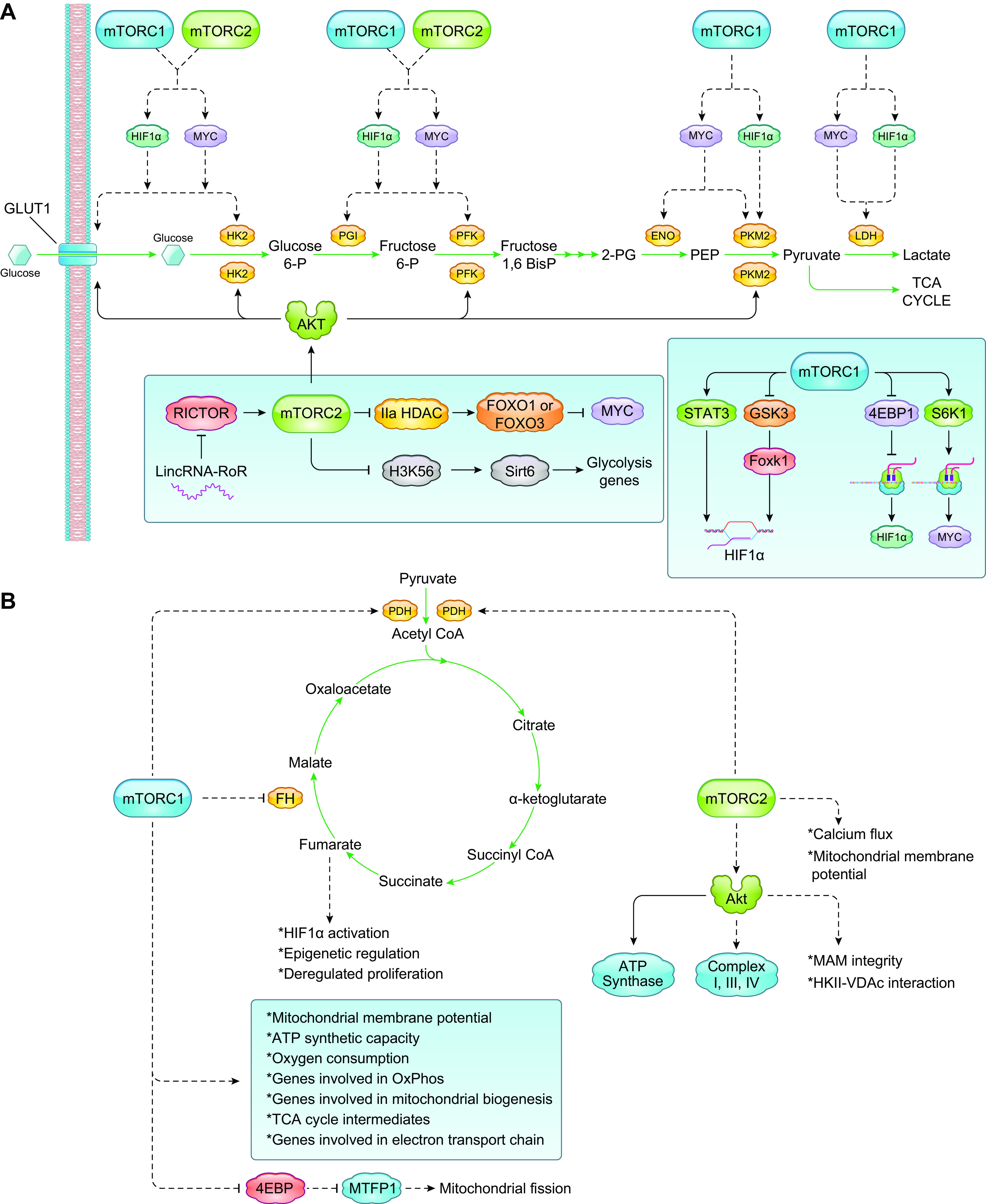
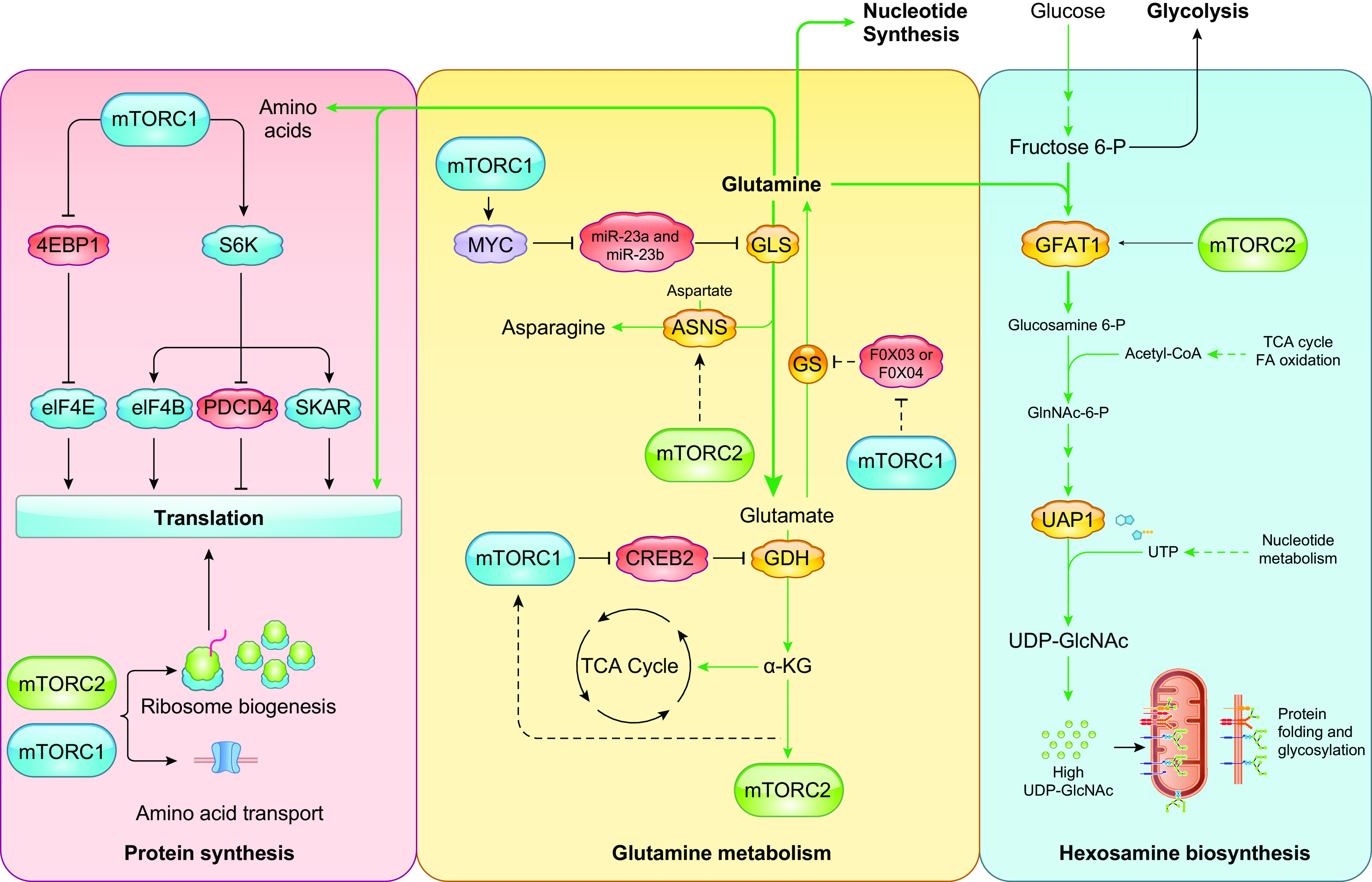
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous