

Assembly and Exploration of a Single Cell Atlas of the Drosophila Larval Ventral Cord. Identification of Rare Cell Types
- PMID: 33600085
- PMCID: PMC7899083
- DOI: 10.1002/cpz1.37
Assembly and Exploration of a Single Cell Atlas of the Drosophila Larval Ventral Cord. Identification of Rare Cell Types
Erratum in
-
Group Correction Statement (Data Availability Statements).Curr Protoc. 2022 Aug;2(8):e552. doi: 10.1002/cpz1.552. Curr Protoc. 2022. PMID: 36005902 Free PMC article. No abstract available.
-
Group Correction Statement (Conflict of Interest Statements).Curr Protoc. 2022 Aug;2(8):e551. doi: 10.1002/cpz1.551. Curr Protoc. 2022. PMID: 36005903 Free PMC article. No abstract available.
Abstract
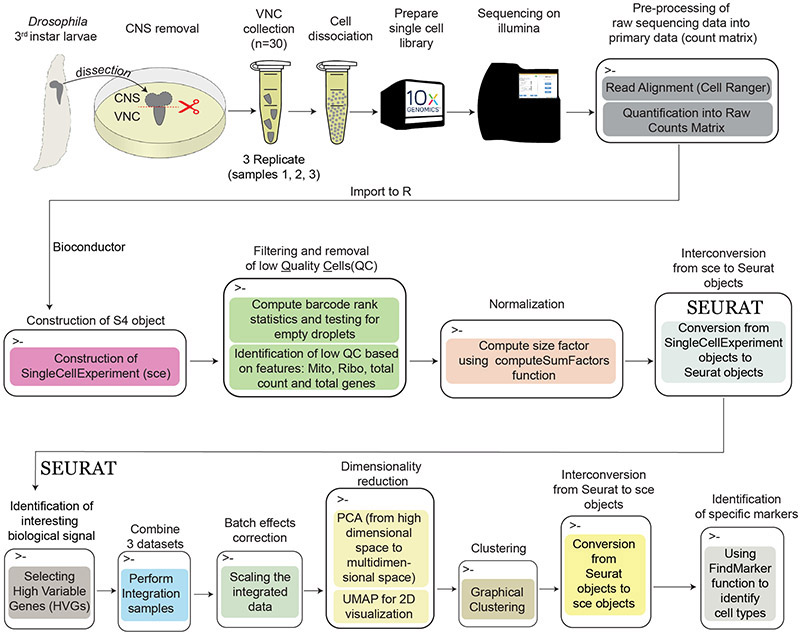
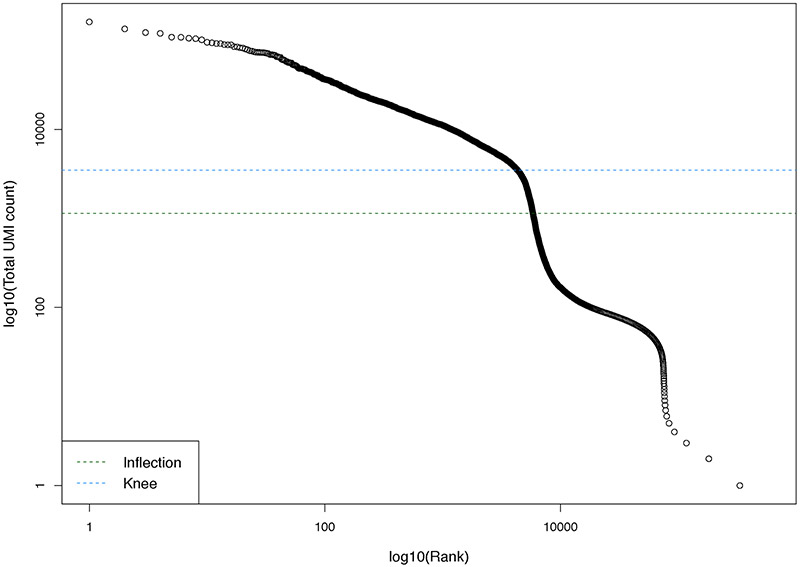
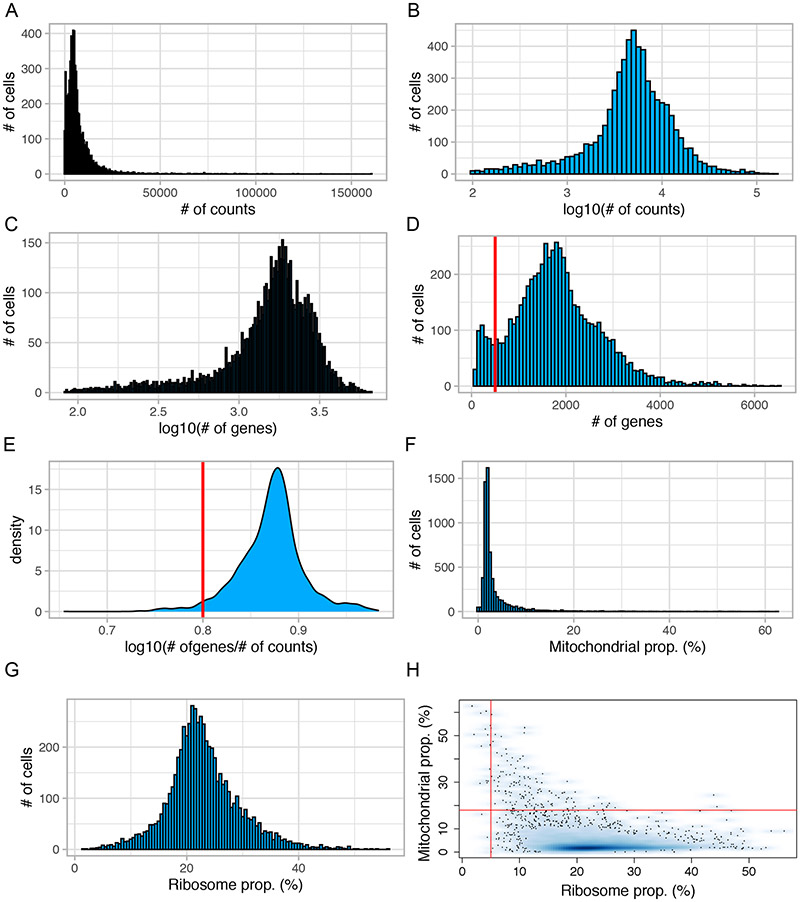
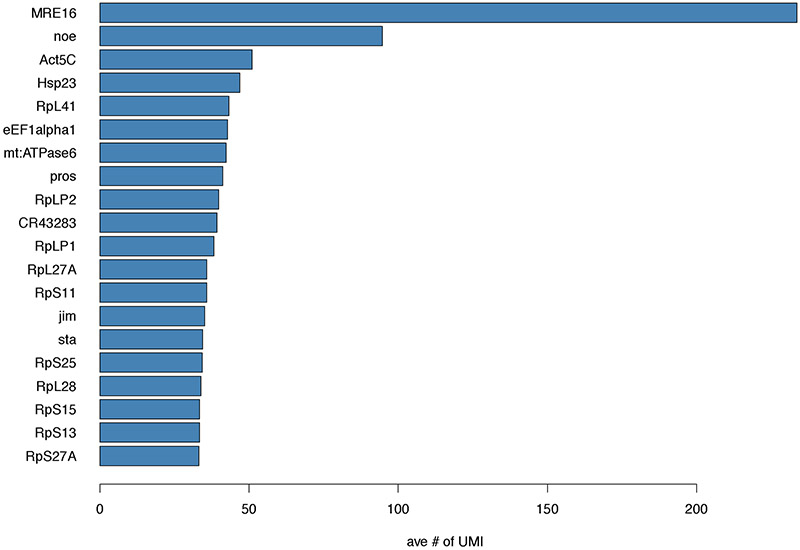
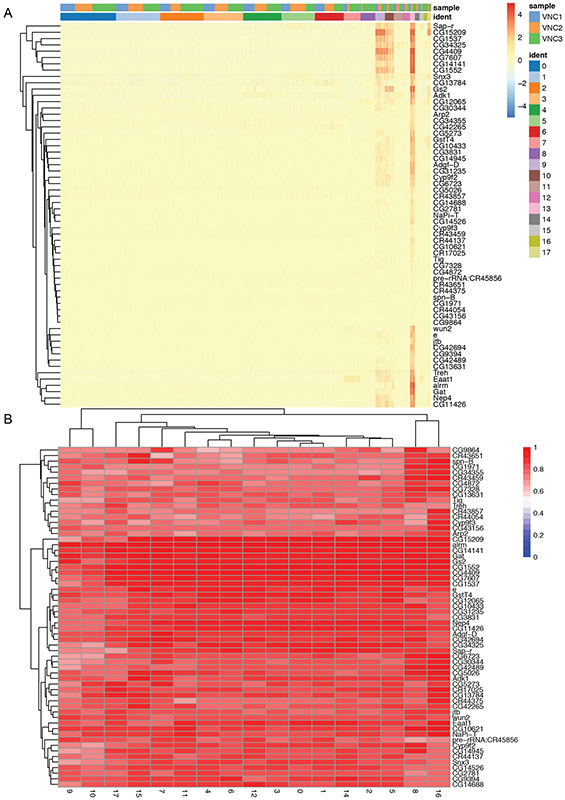
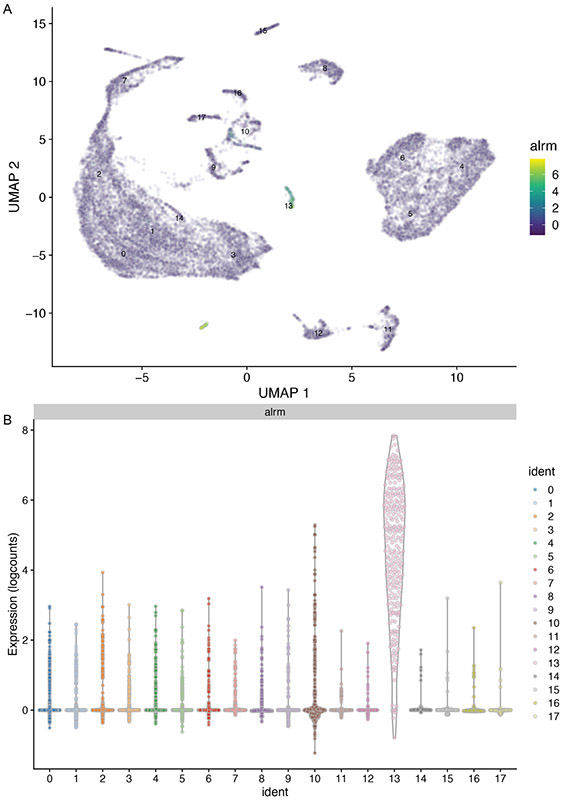
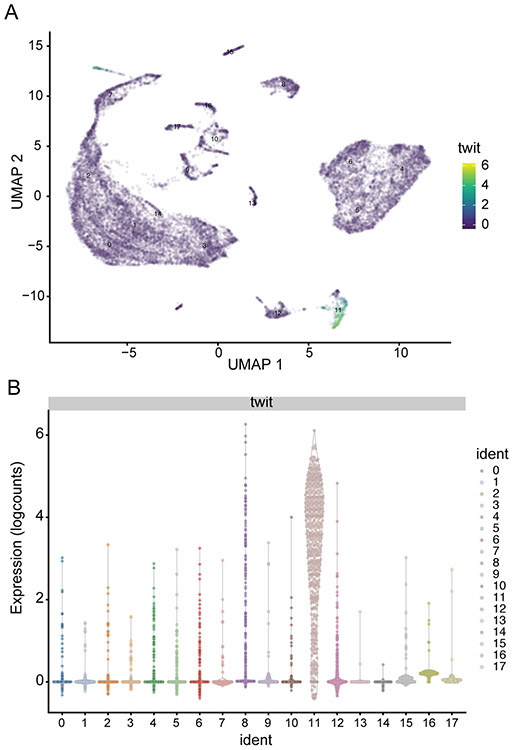
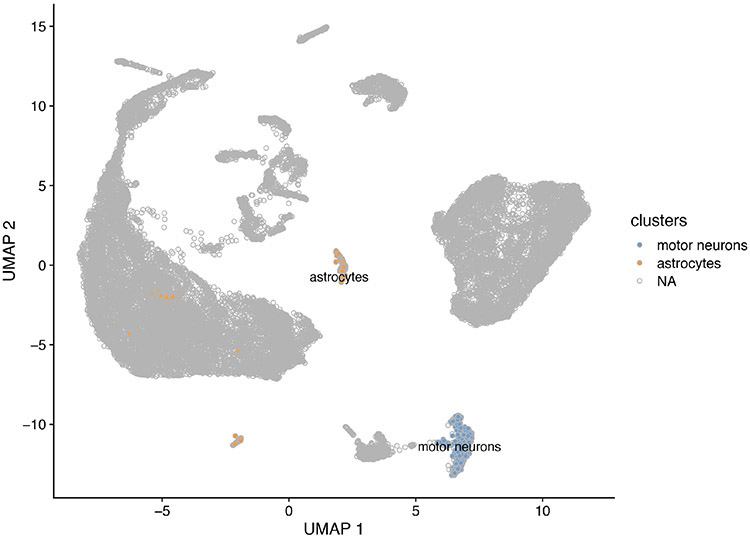
Single-cell RNA sequencing provides a new approach to an old problem: how to study cellular diversity in complex biological systems. This powerful tool has been instrumental in profiling different cell types and investigating, at the single-cell level, cell states, functions, and responses. However, mining these data requires new analytical and statistical methods for high-dimensional analyses that must be customized and adapted to specific goals. Here we present a custom multistage analysis pipeline which integrates modules contained in different R packages to ensure flexible, high-quality RNA-seq data analysis. We describe this workflow step by step, providing the codes, explaining the rationale for each function, and discussing the results and the limitations. We apply this pipeline to analyze different datasets of Drosophila larval ventral cords, identifying and describing rare cell types, such as astrocytes and neuroendocrine cells. This multistage analysis pipeline can be easily implemented by both novice and experienced scientists interested in neuronal and/or cellular diversity beyond the Drosophila model system. © 2021 US Government.
Keywords: R pipeline; cell type identification; clustering; dimensionality reduction; multisample integration; scRNA-seq.
Published 2021. This article is a U.S. Government work and is in the public domain in the USA.
Figures

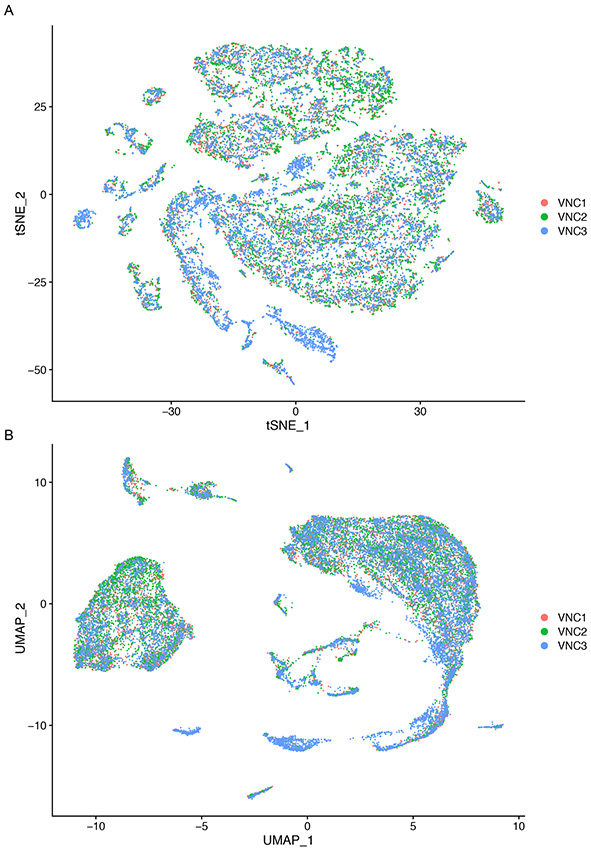
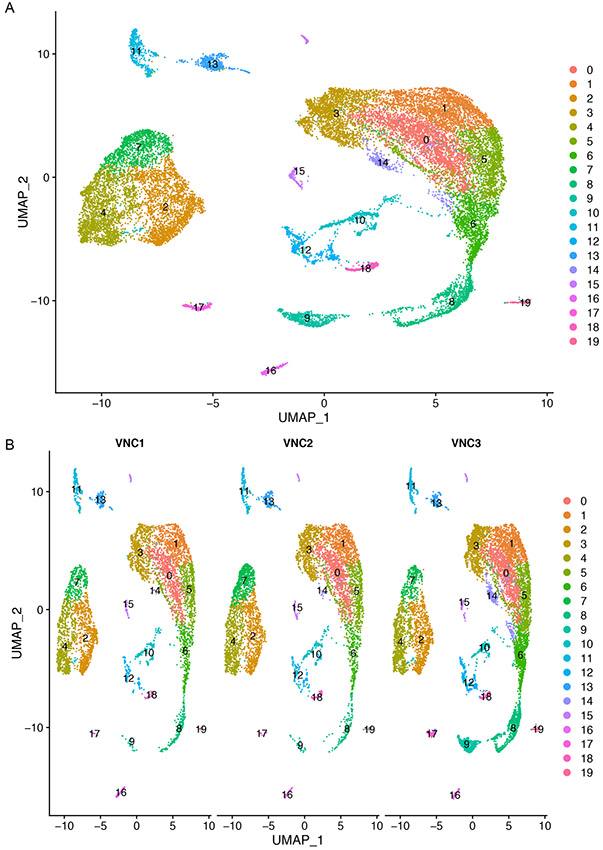
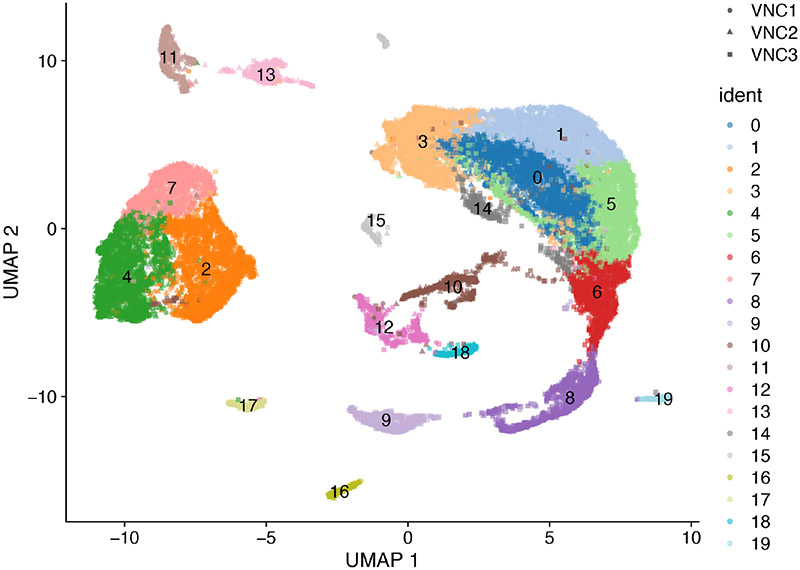
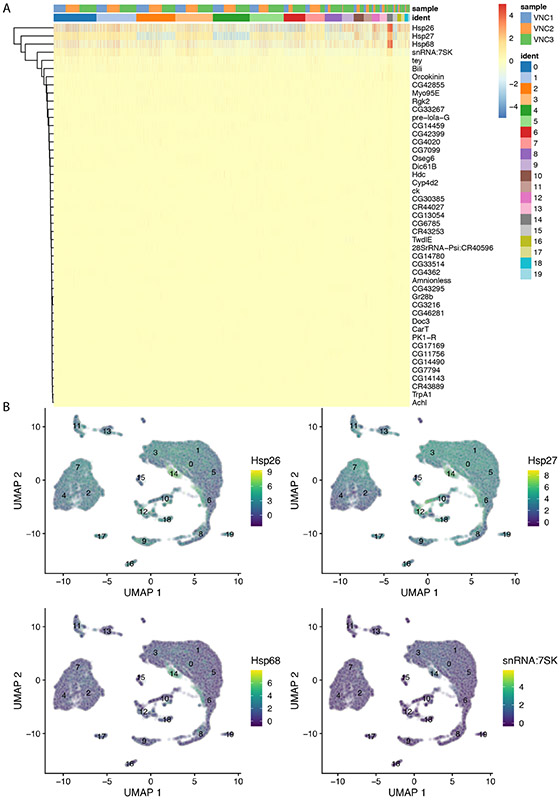
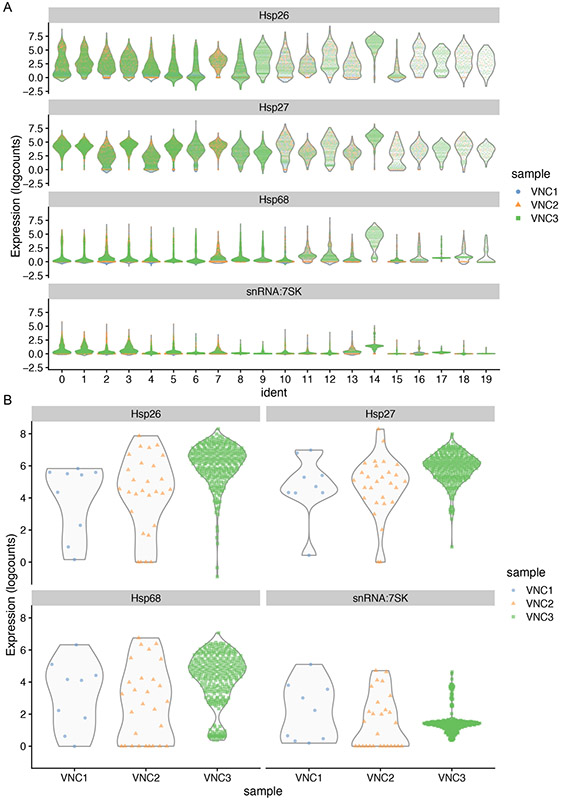
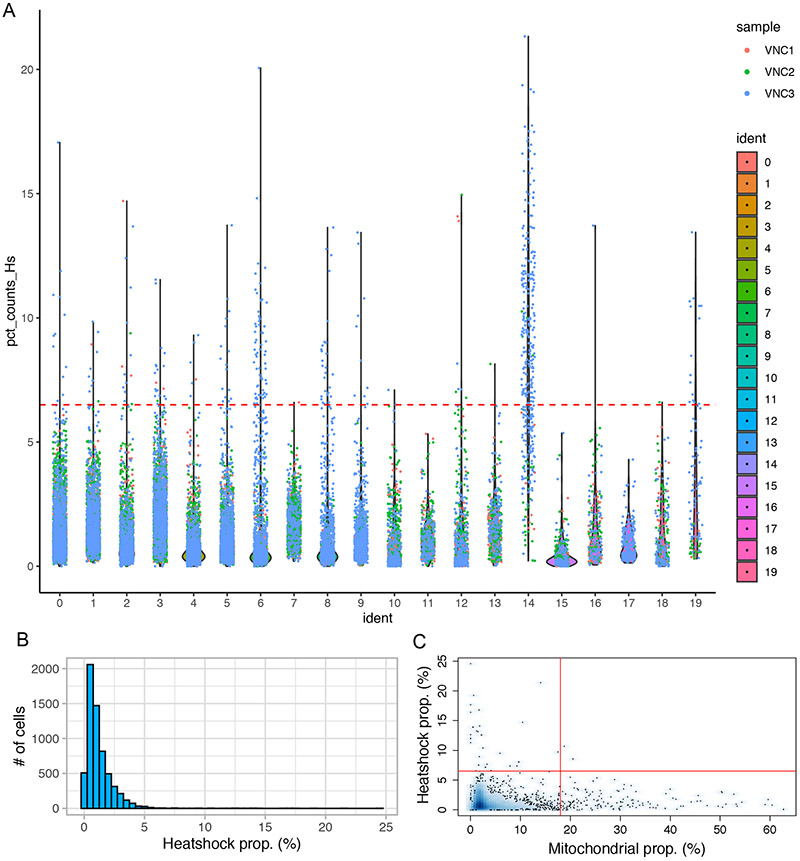
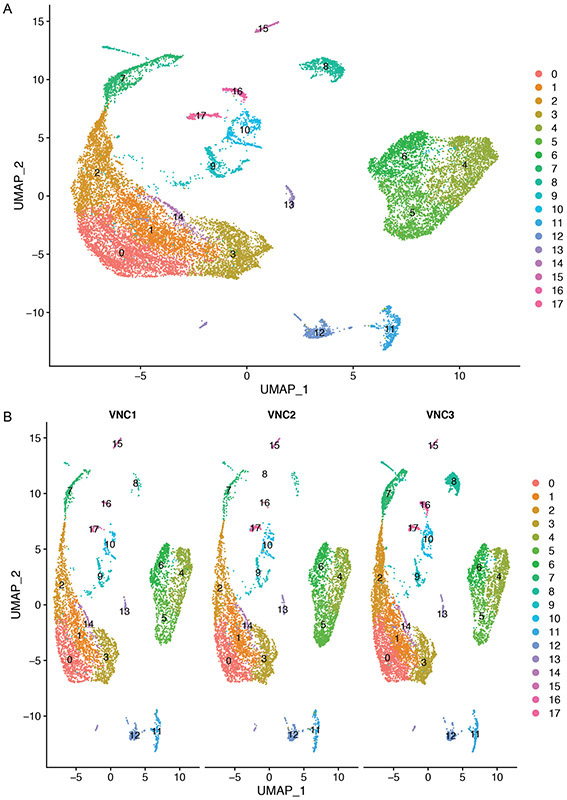
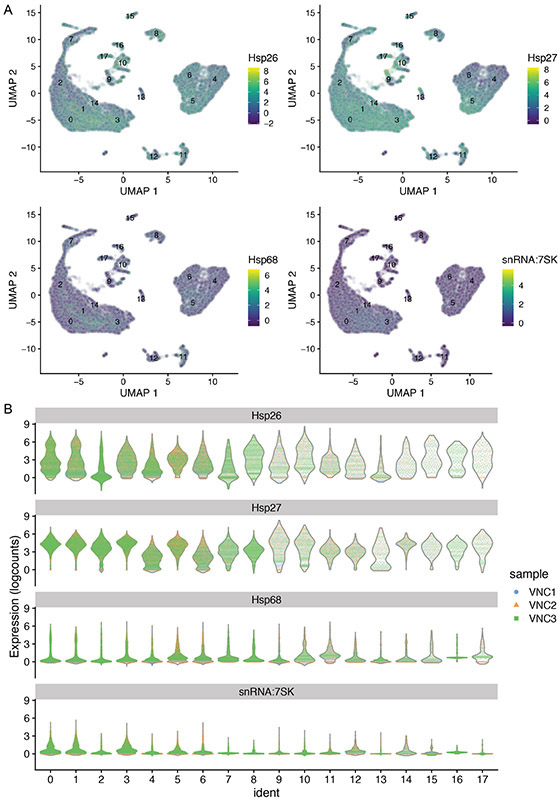
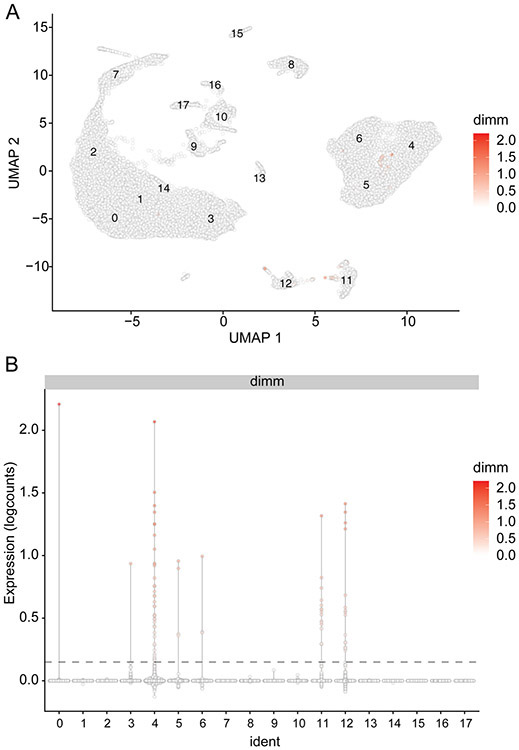
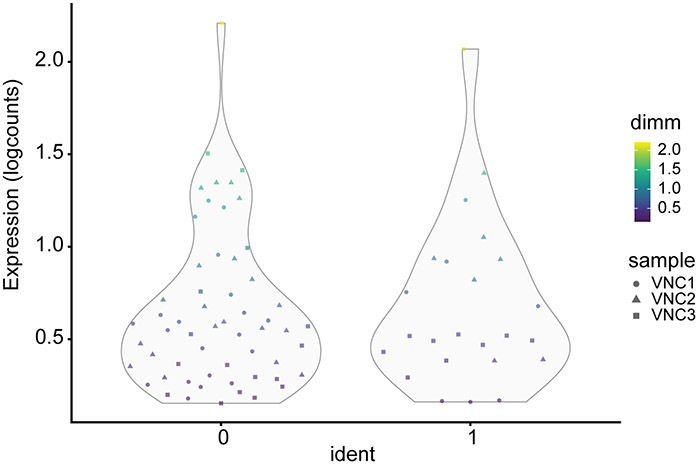
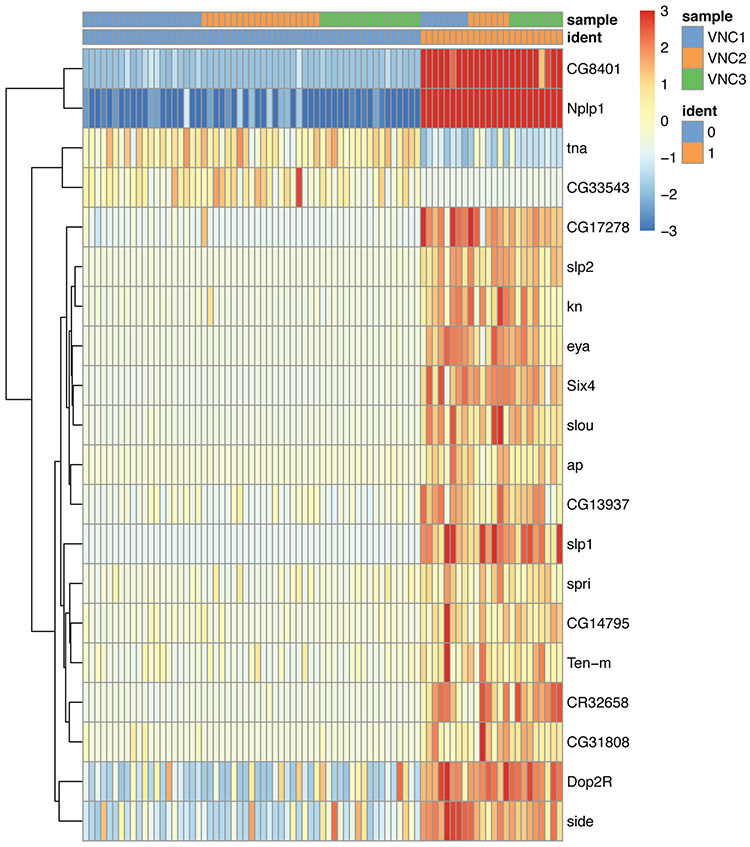
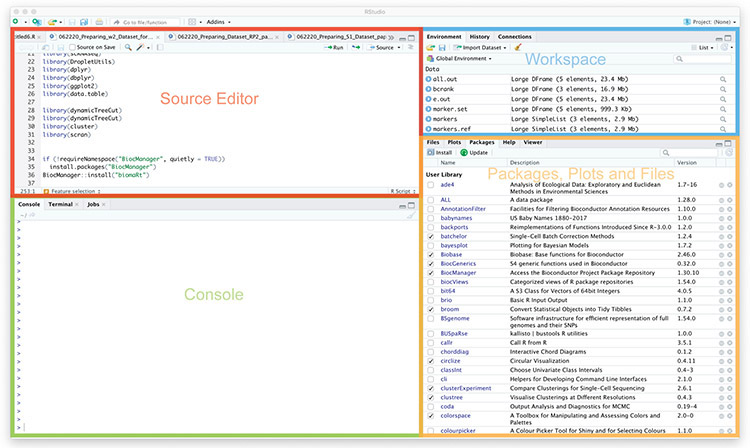
References
-
- Amezquita RA, Lun ATL, Becht E, Carey VJ, Carpp LN, Geistlinger L, Marini F, Rue-Albrecht K, Risso D, Soneson C, et al. (2020). Publisher Correction: Orchestrating single-cell analysis with Bioconductor. Nat Methods 17, 242. - PubMed
-
- Angerer P, Haghverdi L, Buttner M, Theis FJ, Marr C, and Buettner F (2016). destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics 32, 1241–1243. - PubMed
-
- Becht E, McInnes L, Healy J, Dutertre CA, Kwok IWH, Ng LG, Ginhoux F, and Newell EW (2018). Dimensionality reduction for visualizing single-cell data using UMAP. Nat Biotechnol. - PubMed
-
- Brennecke P, Anders S, Kim JK, Kolodziejczyk AA, Zhang X, Proserpio V, Baying B, Benes V, Teichmann SA, Marioni JC, et al. (2013). Accounting for technical noise in single-cell RNA-seq experiments. Nat Methods 10, 1093–1095. - PubMed
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
