

Whole-genome sequencing of African Americans implicates differential genetic architecture in inflammatory bowel disease
- PMID: 33600772
- PMCID: PMC8008495
- DOI: 10.1016/j.ajhg.2021.02.001
Whole-genome sequencing of African Americans implicates differential genetic architecture in inflammatory bowel disease
Abstract
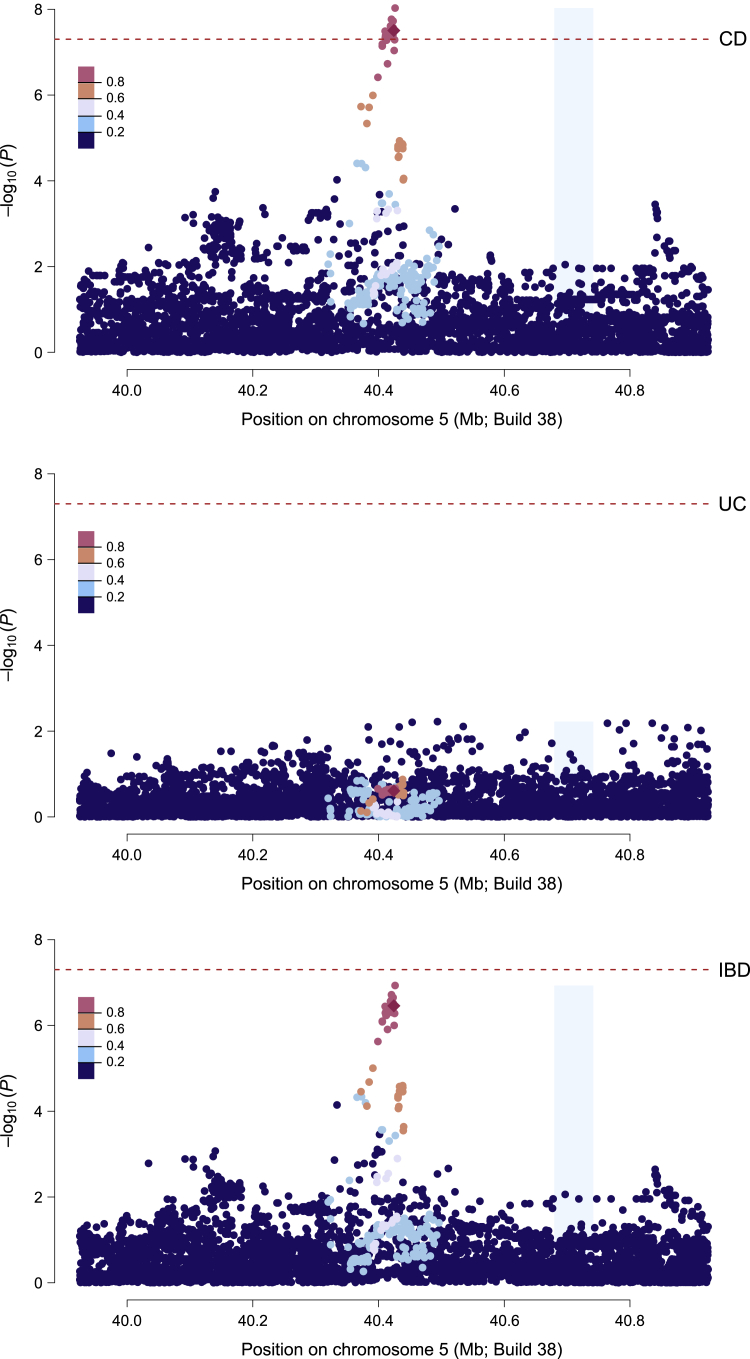
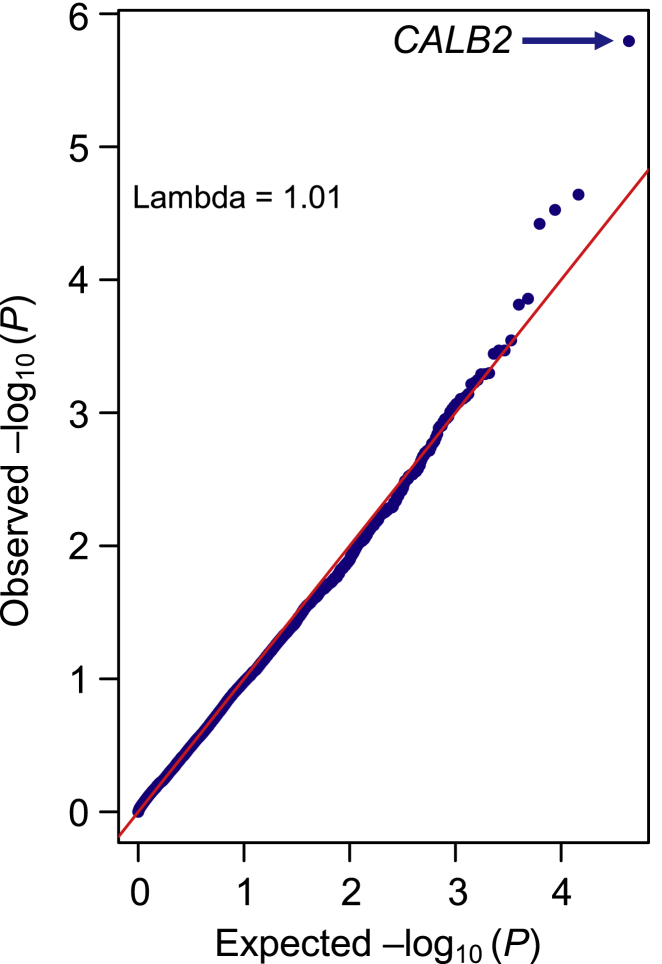
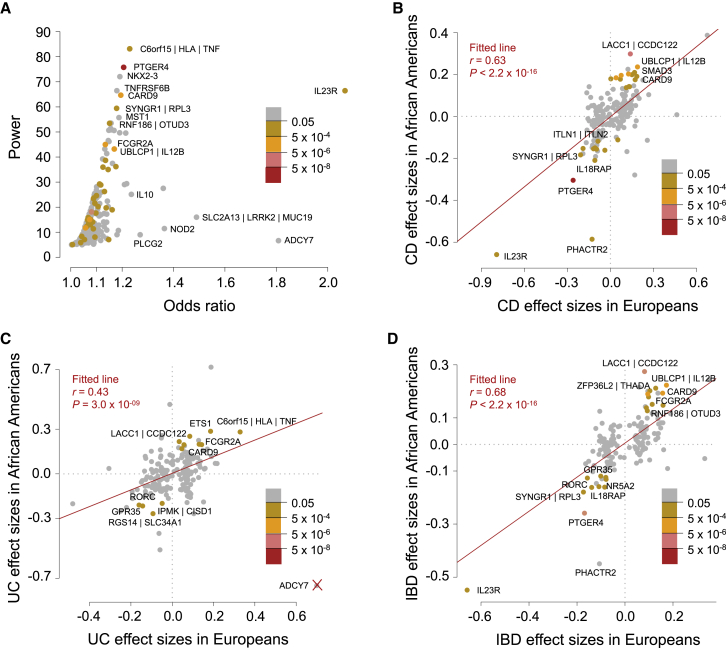
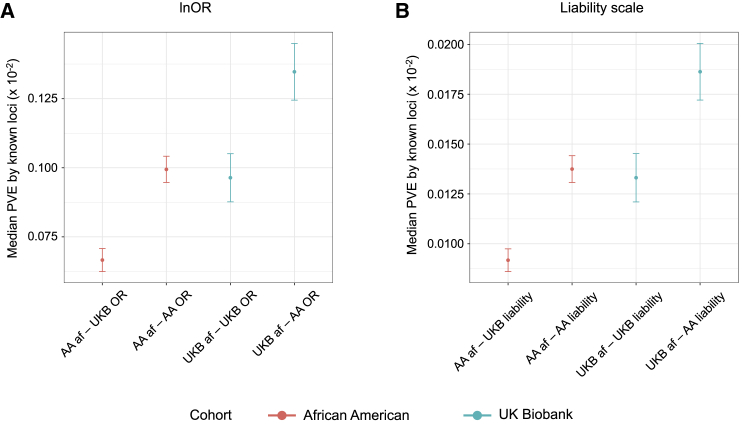
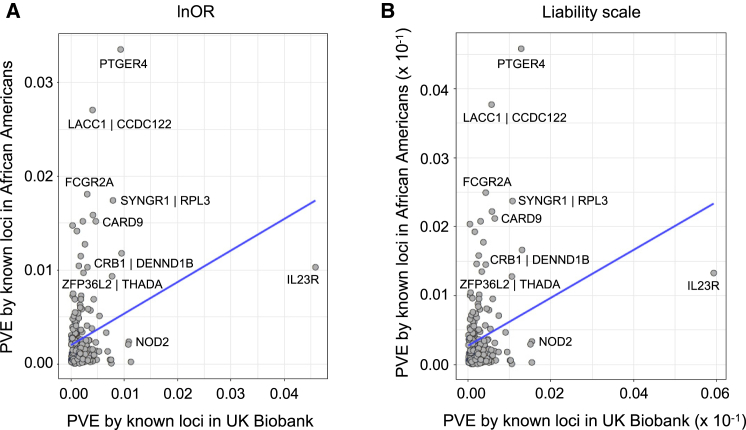
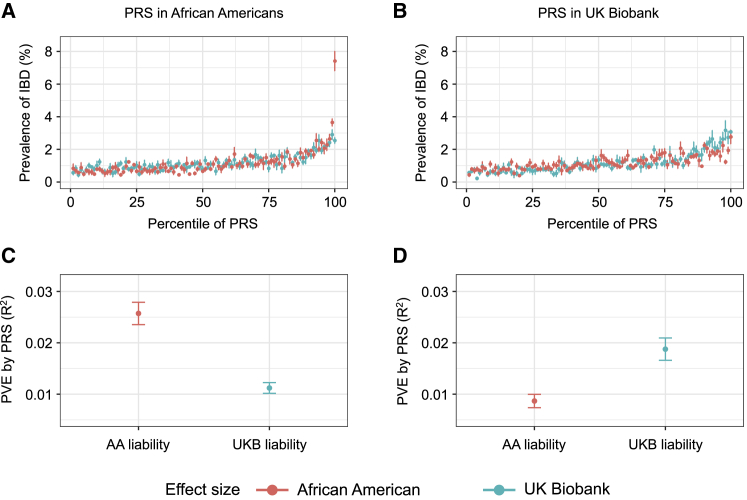
Whether or not populations diverge with respect to the genetic contribution to risk of specific complex diseases is relevant to understanding the evolution of susceptibility and origins of health disparities. Here, we describe a large-scale whole-genome sequencing study of inflammatory bowel disease encompassing 1,774 affected individuals and 1,644 healthy control Americans with African ancestry (African Americans). Although no new loci for inflammatory bowel disease are discovered at genome-wide significance levels, we identify numerous instances of differential effect sizes in combination with divergent allele frequencies. For example, the major effect at PTGER4 fine maps to a single credible interval of 22 SNPs corresponding to one of four independent associations at the locus in European ancestry individuals but with an elevated odds ratio for Crohn disease in African Americans. A rare variant aggregate analysis implicates Ca2+-binding neuro-immunomodulator CALB2 in ulcerative colitis. Highly significant overall overlap of common variant risk for inflammatory bowel disease susceptibility between individuals with African and European ancestries was observed, with 41 of 241 previously known lead variants replicated and overall correlations in effect sizes of 0.68 for combined inflammatory bowel disease. Nevertheless, subtle differences influence the performance of polygenic risk scores, and we show that ancestry-appropriate weights significantly improve polygenic prediction in the highest percentiles of risk. The median amount of variance explained per locus remains the same in African and European cohorts, providing evidence for compensation of effect sizes as allele frequencies diverge, as expected under a highly polygenic model of disease.
Keywords: CALB2; PTGER4; differential genetic architecture; polygenic risk scores; rare variants; trans-ethnic comparative analysis; understudied populations; whole-genome sequencing of African Americans.
Copyright © 2021 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
-
- Jostins L., Ripke S., Weersma R.K., Duerr R.H., McGovern D.P., Hui K.Y., Lee J.C., Schumm L.P., Sharma Y., Anderson C.A., International IBD Genetics Consortium (IIBDGC) Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. - PMC - PubMed
-
- Liu J.Z., van Sommeren S., Huang H., Ng S.C., Alberts R., Takahashi A., Ripke S., Lee J.C., Jostins L., Shah T., International Multiple Sclerosis Genetics Consortium. International IBD Genetics Consortium Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015;47:979–986. - PMC - PubMed
-
- Long T., Hicks M., Yu H.C., Biggs W.H., Kirkness E.F., Menni C., Zierer J., Small K.S., Mangino M., Messier H. Whole-genome sequencing identifies common-to-rare variants associated with human blood metabolites. Nat. Genet. 2017;49:568–578. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U54 DE023789/DE/NIDCR NIH HHS/United States
- P30 DK042086/DK/NIDDK NIH HHS/United States
- U24 DK062429/DK/NIDDK NIH HHS/United States
- U01 DK062432/DK/NIDDK NIH HHS/United States
- U01 DK062429/DK/NIDDK NIH HHS/United States
- U01 DK062422/DK/NIDDK NIH HHS/United States
- U01 DK062423/DK/NIDDK NIH HHS/United States
- U54 HG003067/HG/NHGRI NIH HHS/United States
- U01 DK062413/DK/NIDDK NIH HHS/United States
- UM1 HG008895/HG/NHGRI NIH HHS/United States
- P30 DK043351/DK/NIDDK NIH HHS/United States
- U01 AI067068/AI/NIAID NIH HHS/United States
- P01 DK046763/DK/NIDDK NIH HHS/United States
- U01 DK062420/DK/NIDDK NIH HHS/United States
- U01 DK062431/DK/NIDDK NIH HHS/United States
- R01 DK087694/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
