

POLRMT mutations impair mitochondrial transcription causing neurological disease
- PMID: 33602924
- PMCID: PMC7893070
- DOI: 10.1038/s41467-021-21279-0
POLRMT mutations impair mitochondrial transcription causing neurological disease
Abstract
While >300 disease-causing variants have been identified in the mitochondrial DNA (mtDNA) polymerase γ, no mitochondrial phenotypes have been associated with POLRMT, the RNA polymerase responsible for transcription of the mitochondrial genome. Here, we characterise the clinical and molecular nature of POLRMT variants in eight individuals from seven unrelated families. Patients present with global developmental delay, hypotonia, short stature, and speech/intellectual disability in childhood; one subject displayed an indolent progressive external ophthalmoplegia phenotype. Massive parallel sequencing of all subjects identifies recessive and dominant variants in the POLRMT gene. Patient fibroblasts have a defect in mitochondrial mRNA synthesis, but no mtDNA deletions or copy number abnormalities. The in vitro characterisation of the recombinant POLRMT mutants reveals variable, but deleterious effects on mitochondrial transcription. Together, our in vivo and in vitro functional studies of POLRMT variants establish defective mitochondrial transcription as an important disease mechanism.
Conflict of interest statement
The authors declare no competing interests.
Figures
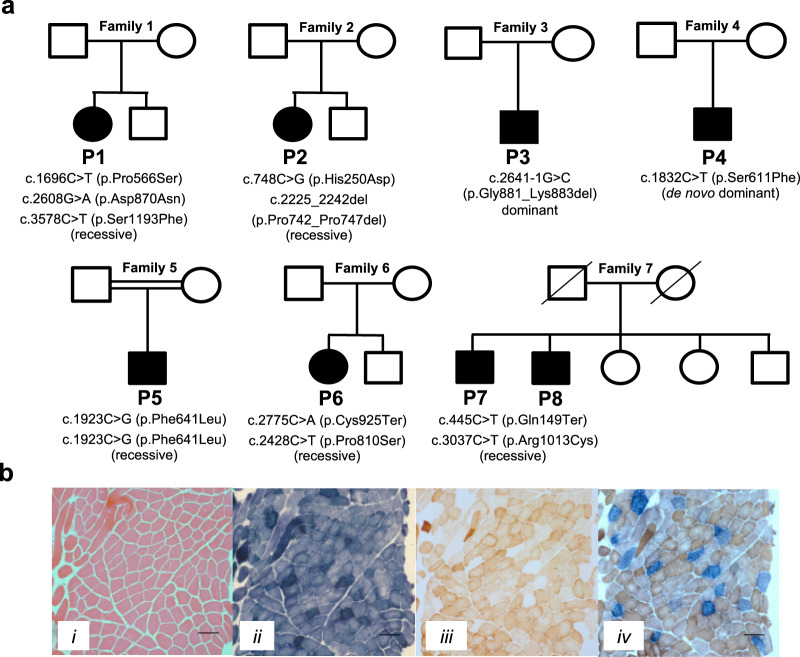
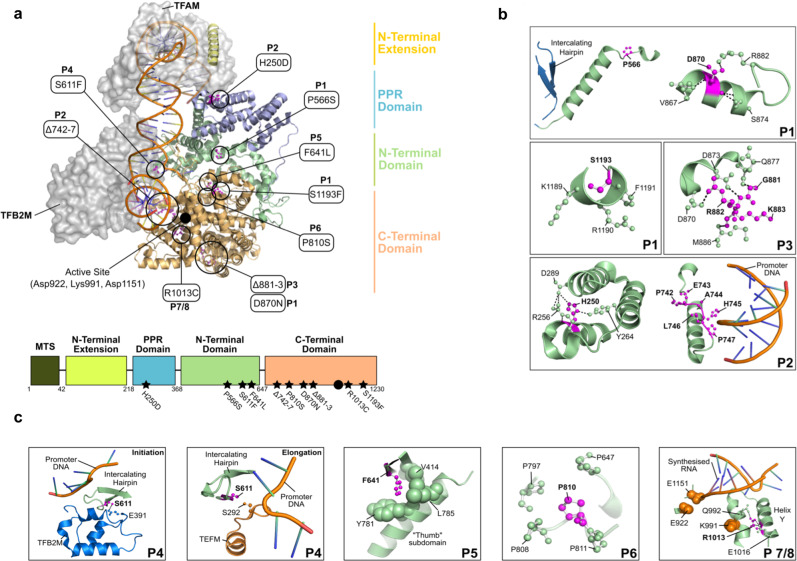
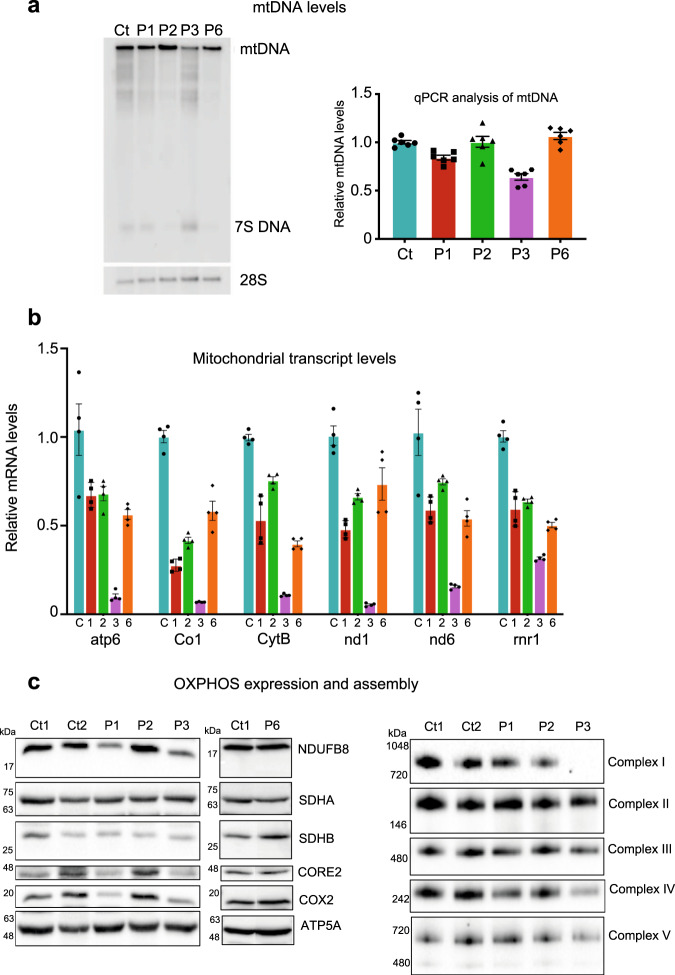
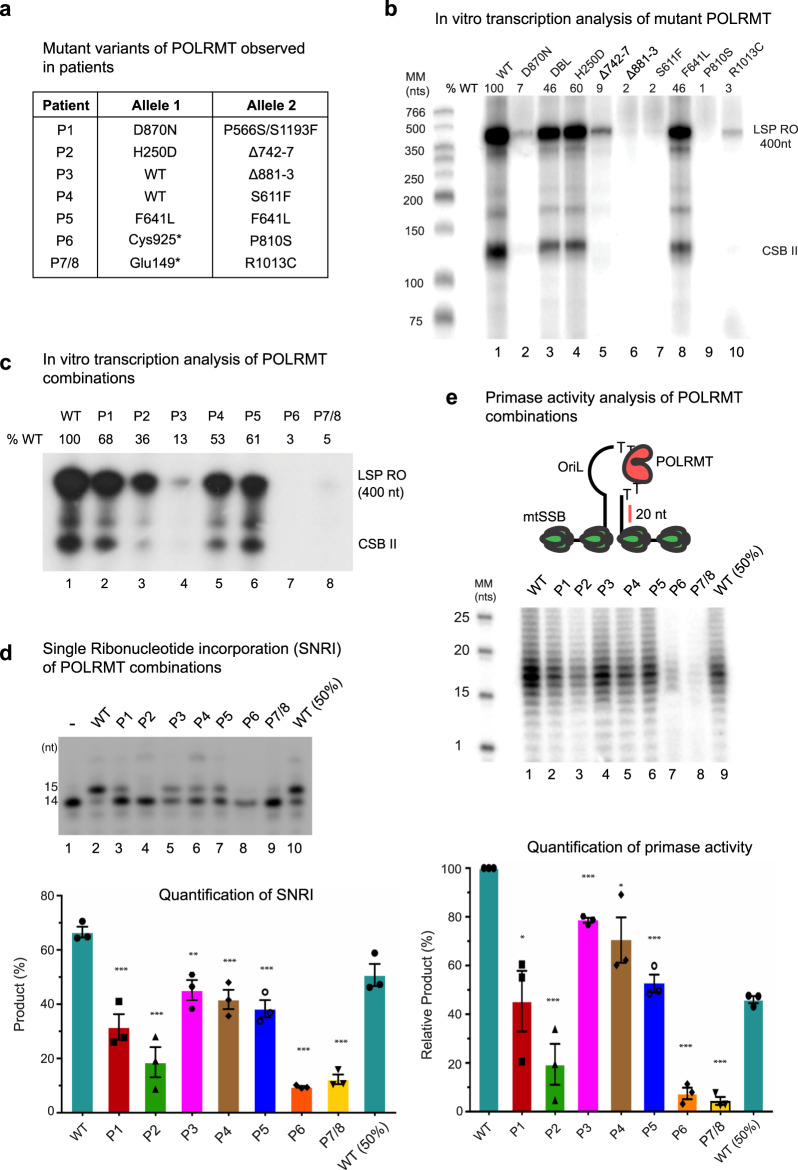
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
