

Temporal association between human upper respiratory and gut bacterial microbiomes during the course of COVID-19 in adults
- PMID: 33603076
- PMCID: PMC7893062
- DOI: 10.1038/s42003-021-01796-w
Temporal association between human upper respiratory and gut bacterial microbiomes during the course of COVID-19 in adults
Abstract
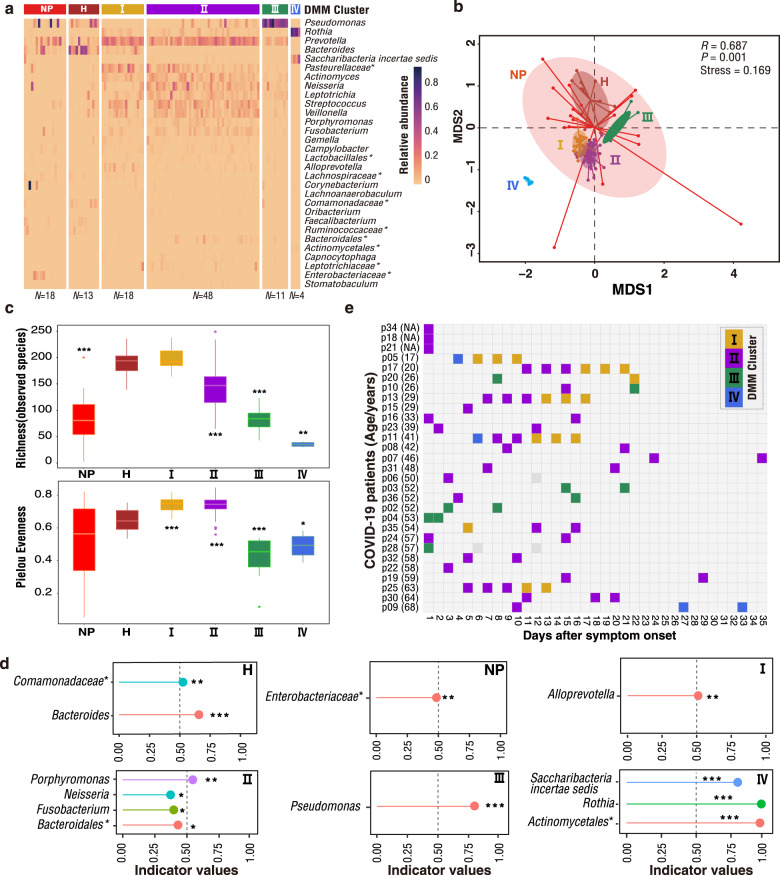
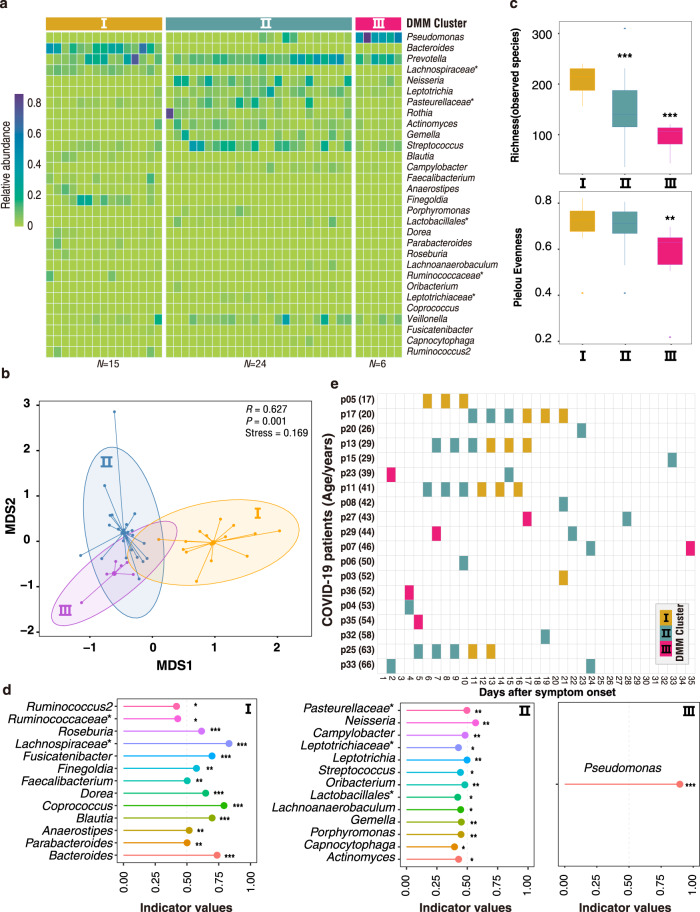
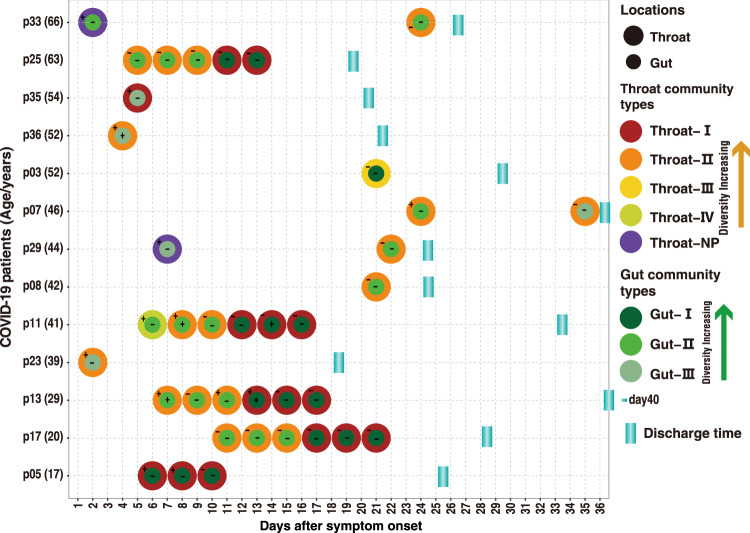
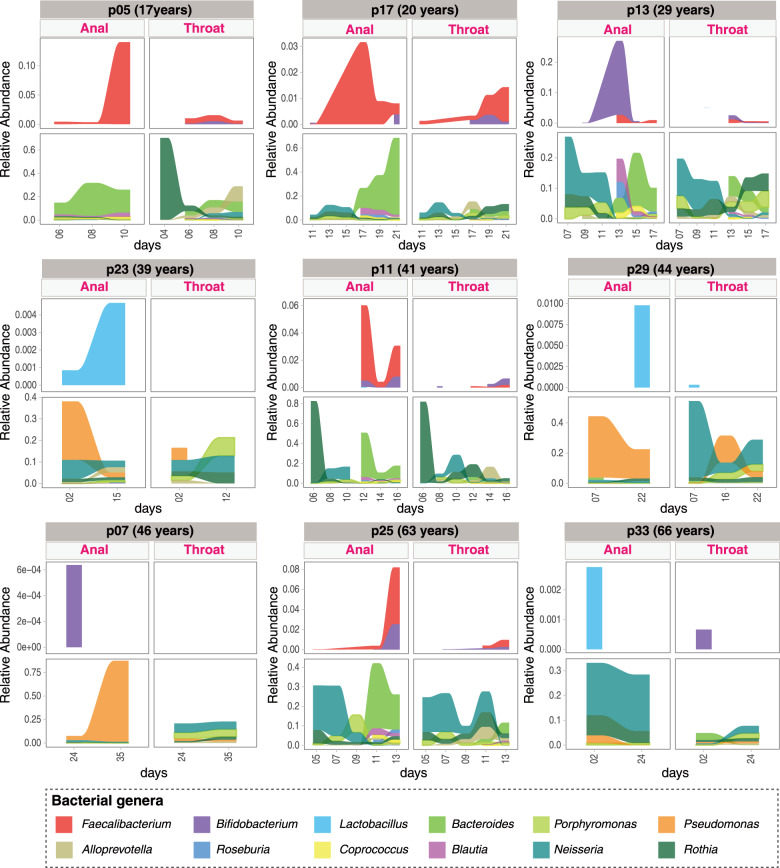
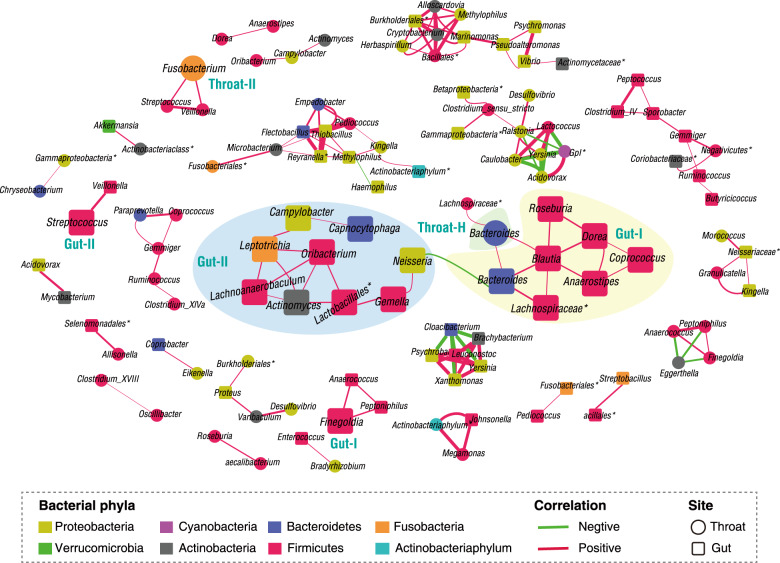
SARS-CoV-2 is the cause of COVID-19. It infects multiple organs including the respiratory tract and gut. Dynamic changes of regional microbiomes in infected adults are largely unknown. Here, we performed longitudinal analyses of throat and anal swabs from 35 COVID-19 and 19 healthy adult controls, as well as 10 non-COVID-19 patients with other diseases, by 16 S rRNA gene sequencing. The results showed a partitioning of the patients into 3-4 categories based on microbial community types (I-IV) in both sites. The bacterial diversity was lower in COVID-19 patients than healthy controls and decreased gradually from community type I to III/IV. Although the dynamic change of microbiome was complex during COVID-19, a synchronous restoration of both the upper respiratory and gut microbiomes from early dysbiosis towards late more diverse status was observed in 6/8 mild COVID-19 adult patients. These findings reveal previously unknown interactions between upper respiratory and gut microbiomes during COVID-19.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
