

Genetic engineering of T cells for immunotherapy
- PMID: 33603158
- PMCID: PMC8217325
- DOI: 10.1038/s41576-021-00329-9
Genetic engineering of T cells for immunotherapy
Abstract
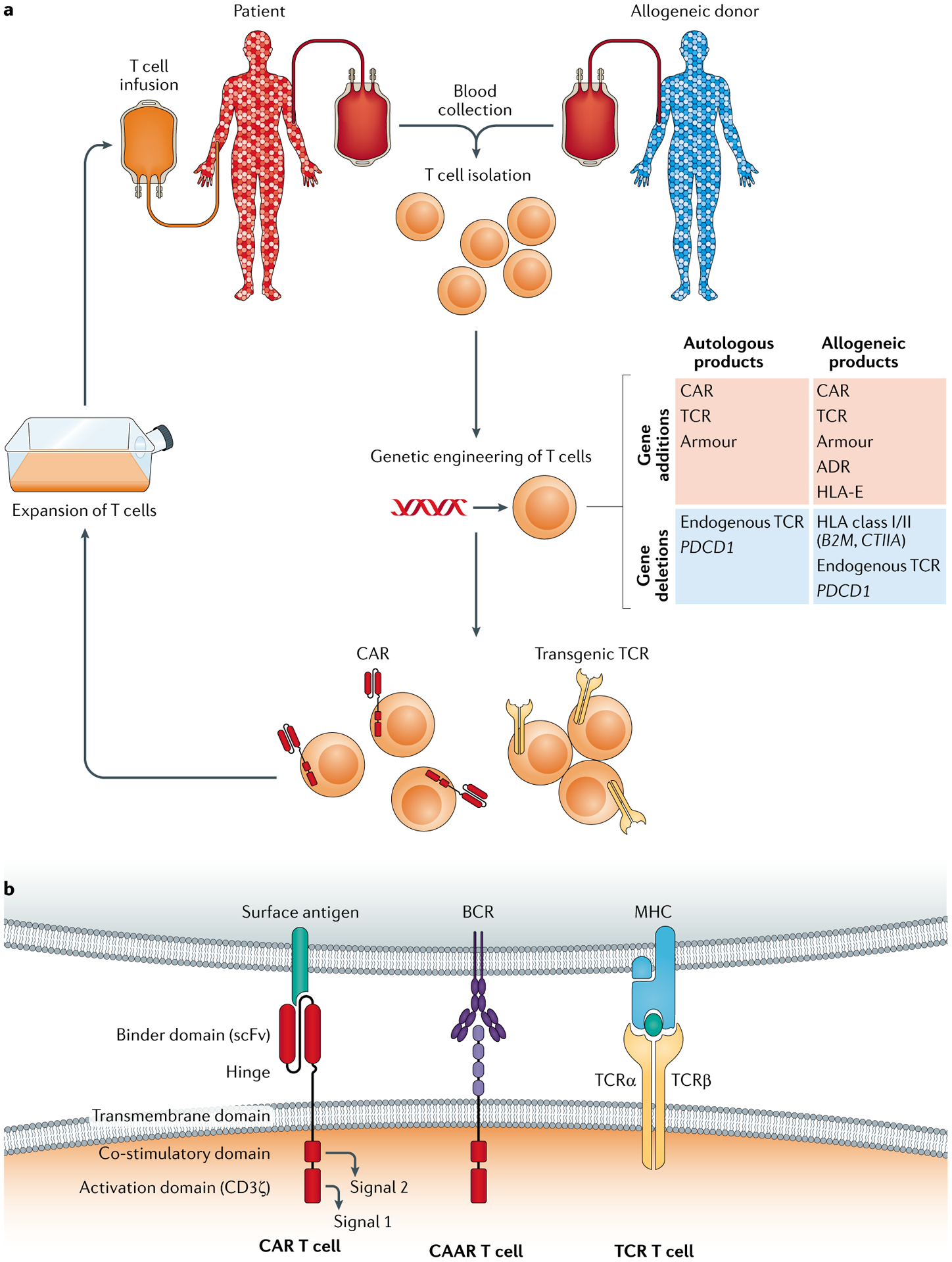
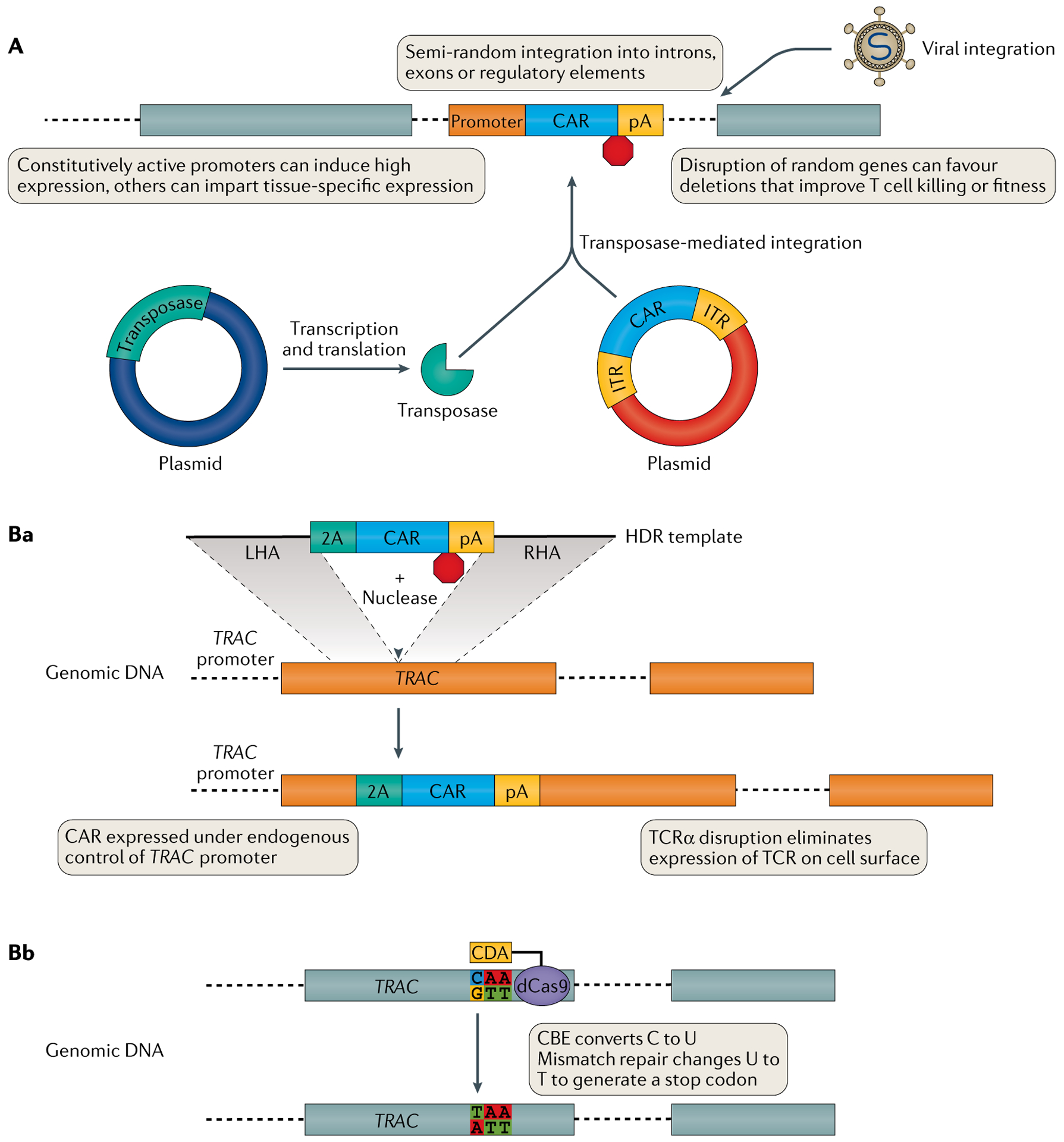
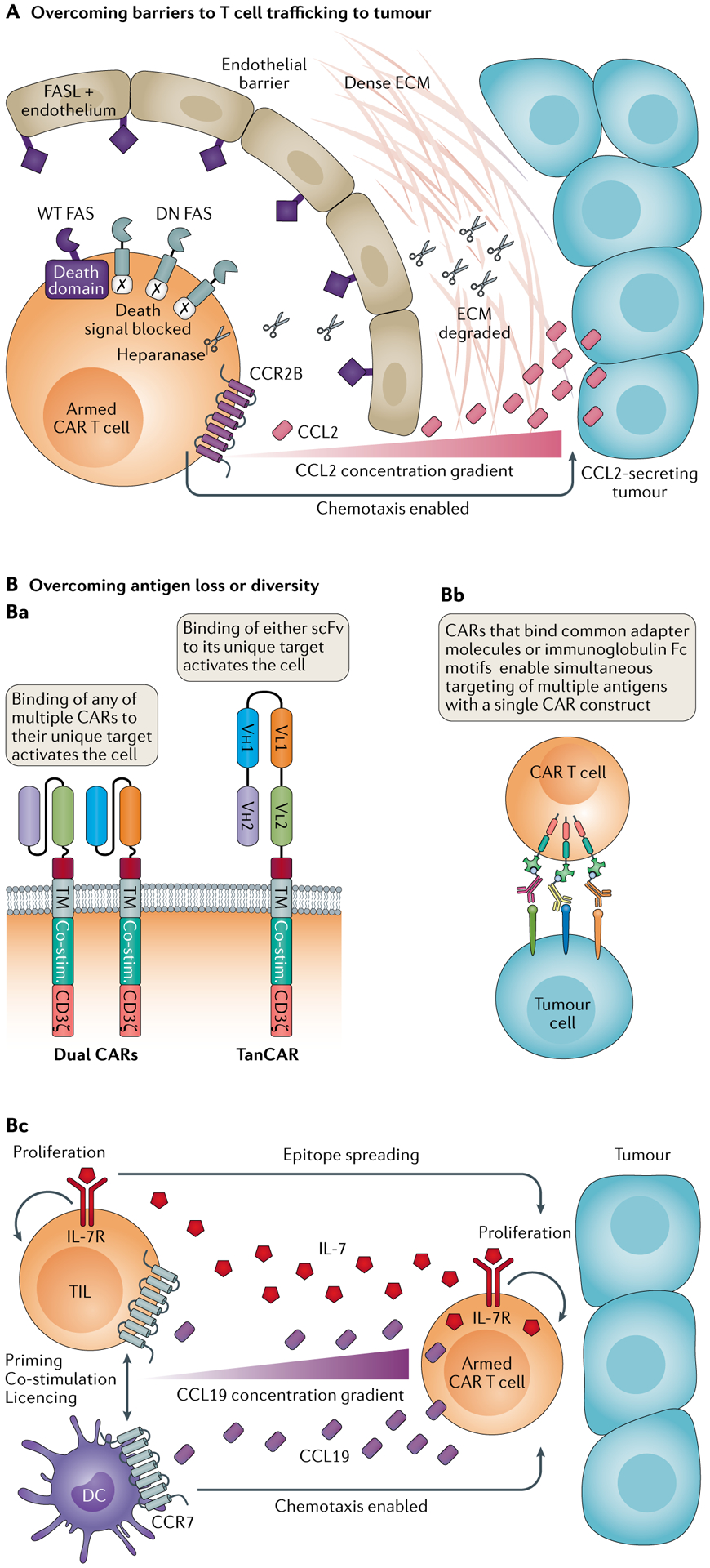
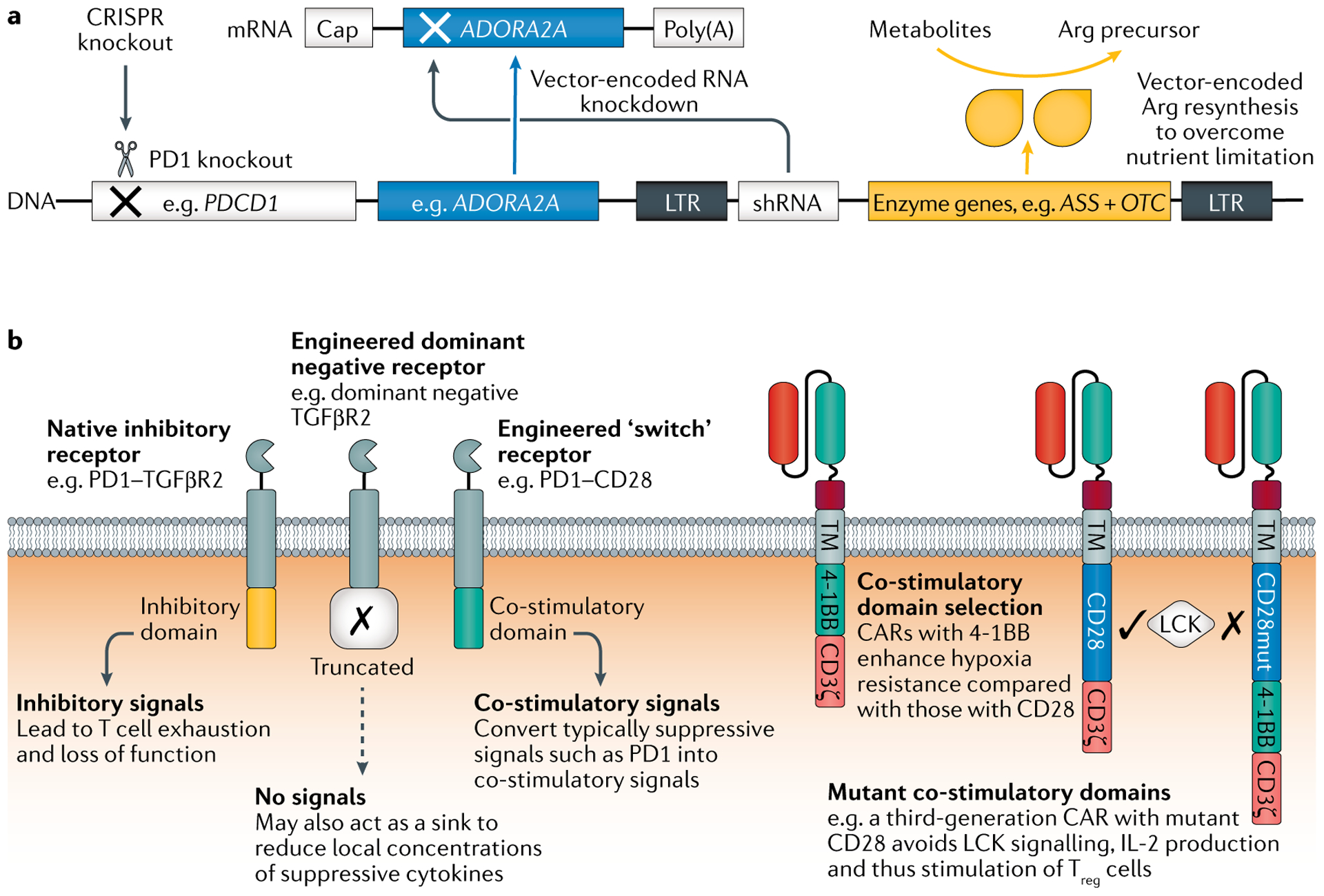
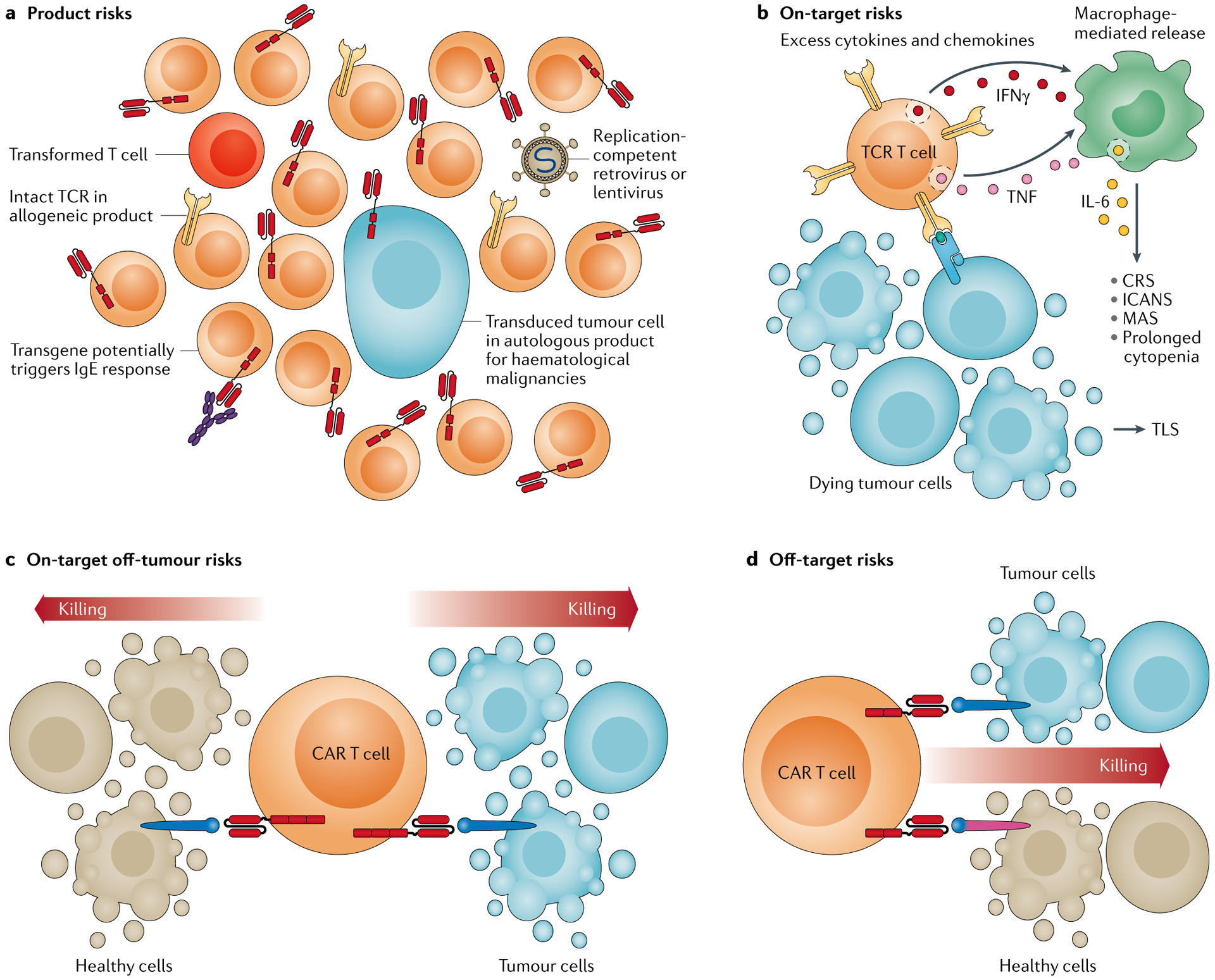
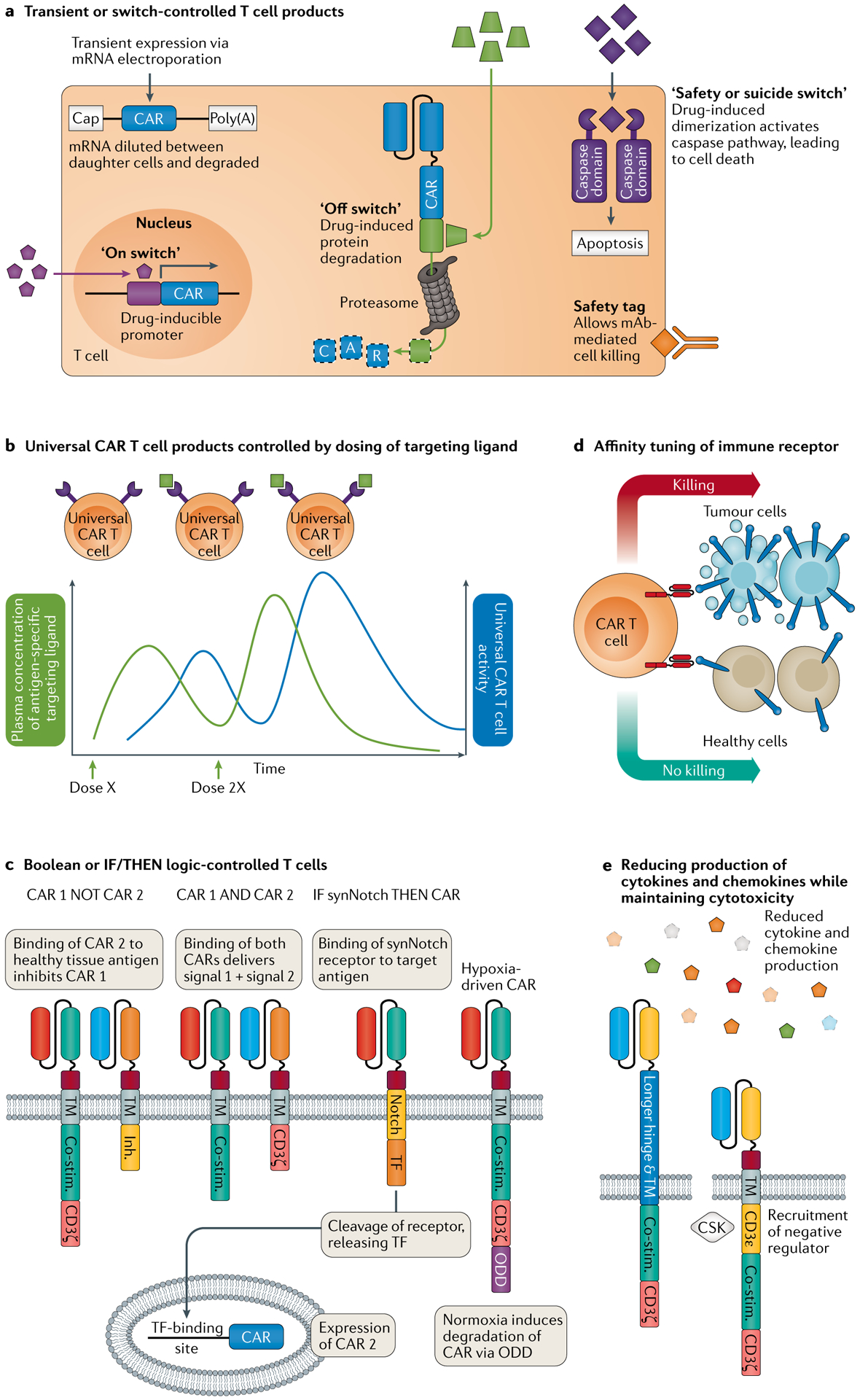
Genetically engineered T cell immunotherapies have provided remarkable clinical success to treat B cell acute lymphoblastic leukaemia by harnessing a patient's own T cells to kill cancer, and these approaches have the potential to provide therapeutic benefit for numerous other cancers, infectious diseases and autoimmunity. By introduction of either a transgenic T cell receptor or a chimeric antigen receptor, T cells can be programmed to target cancer cells. However, initial studies have made it clear that the field will need to implement more complex levels of genetic regulation of engineered T cells to ensure both safety and efficacy. Here, we review the principles by which our knowledge of genetics and genome engineering will drive the next generation of adoptive T cell therapies.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
