

Base-resolution models of transcription-factor binding reveal soft motif syntax
- PMID: 33603233
- PMCID: PMC8812996
- DOI: 10.1038/s41588-021-00782-6
Base-resolution models of transcription-factor binding reveal soft motif syntax
Abstract
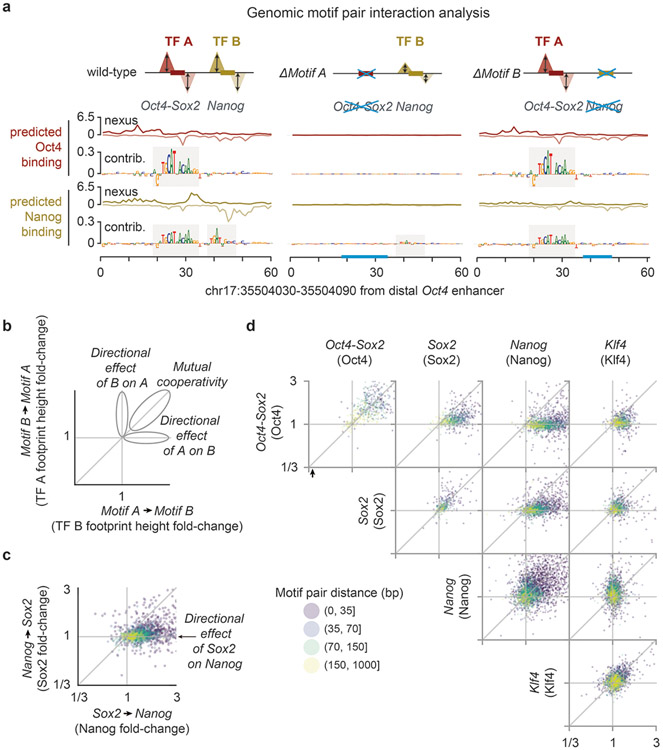
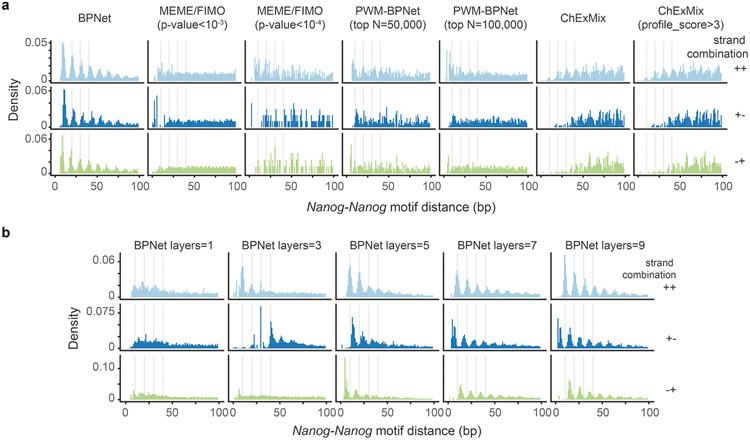
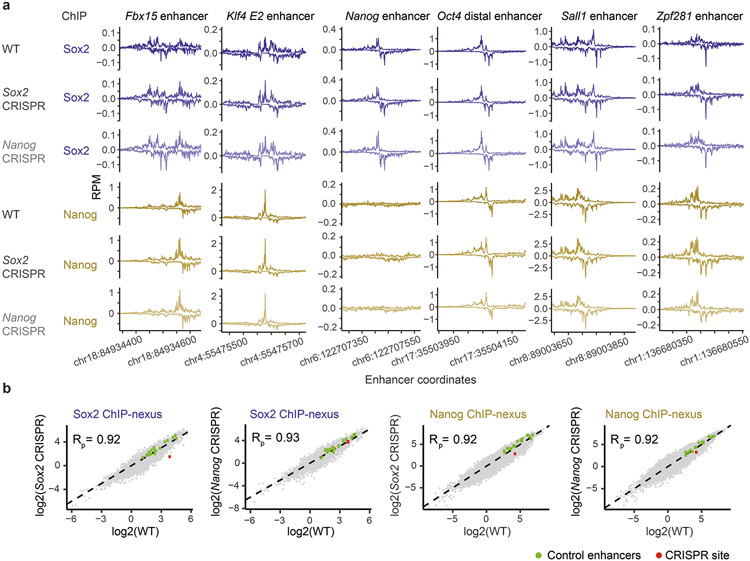
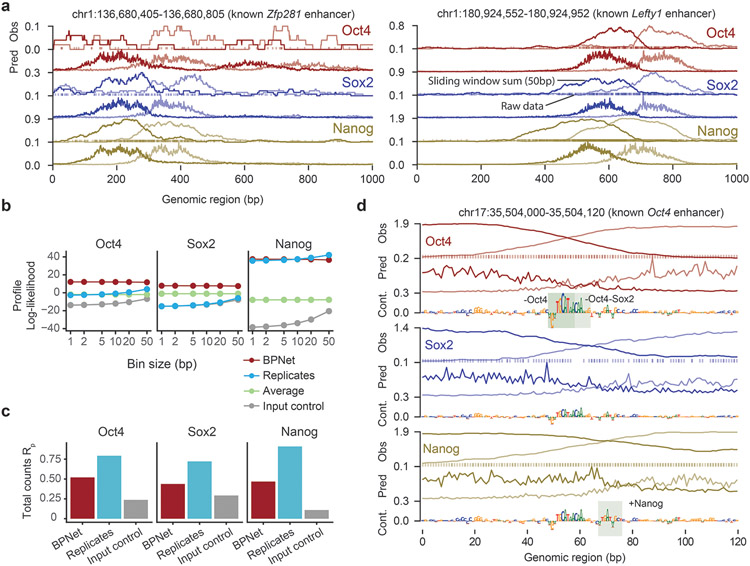
The arrangement (syntax) of transcription factor (TF) binding motifs is an important part of the cis-regulatory code, yet remains elusive. We introduce a deep learning model, BPNet, that uses DNA sequence to predict base-resolution chromatin immunoprecipitation (ChIP)-nexus binding profiles of pluripotency TFs. We develop interpretation tools to learn predictive motif representations and identify soft syntax rules for cooperative TF binding interactions. Strikingly, Nanog preferentially binds with helical periodicity, and TFs often cooperate in a directional manner, which we validate using clustered regularly interspaced short palindromic repeat (CRISPR)-induced point mutations. Our model represents a powerful general approach to uncover the motifs and syntax of cis-regulatory sequences in genomics data.
Conflict of interest statement
Conflict of Interests statement
J.Z. owns a patent on ChIP-nexus (Patent No. 10287628). All other authors declare no competing interests.
Figures
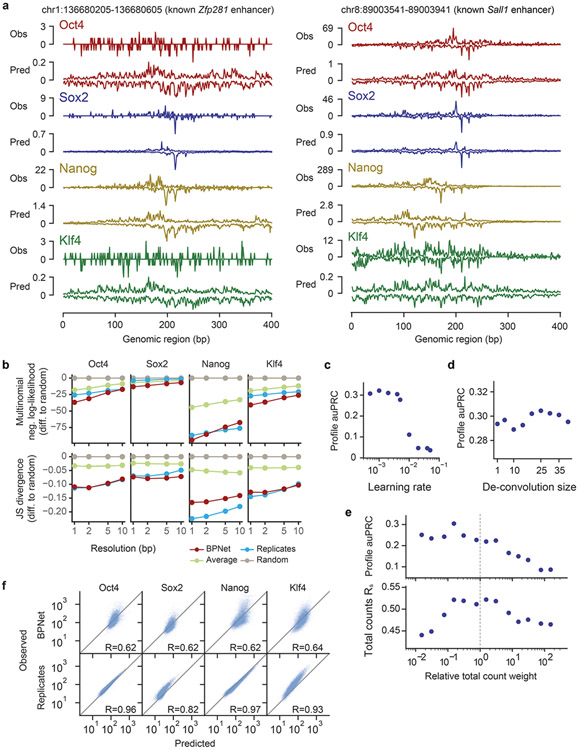

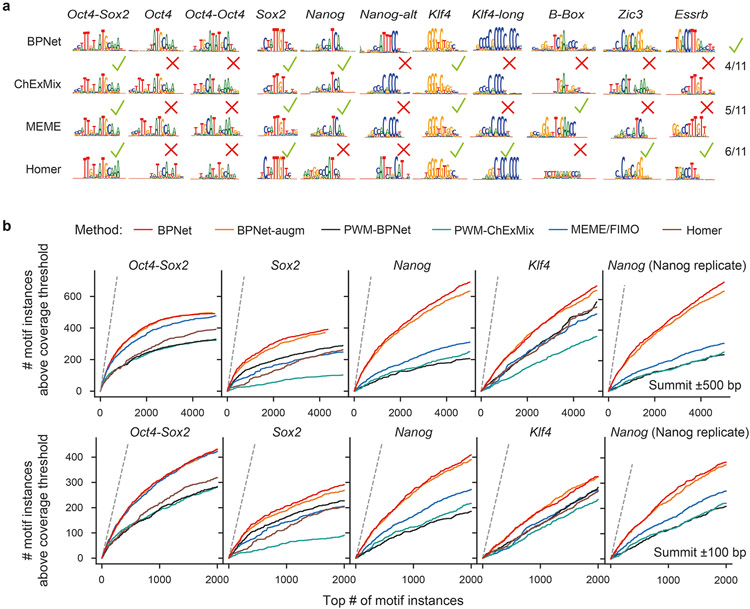
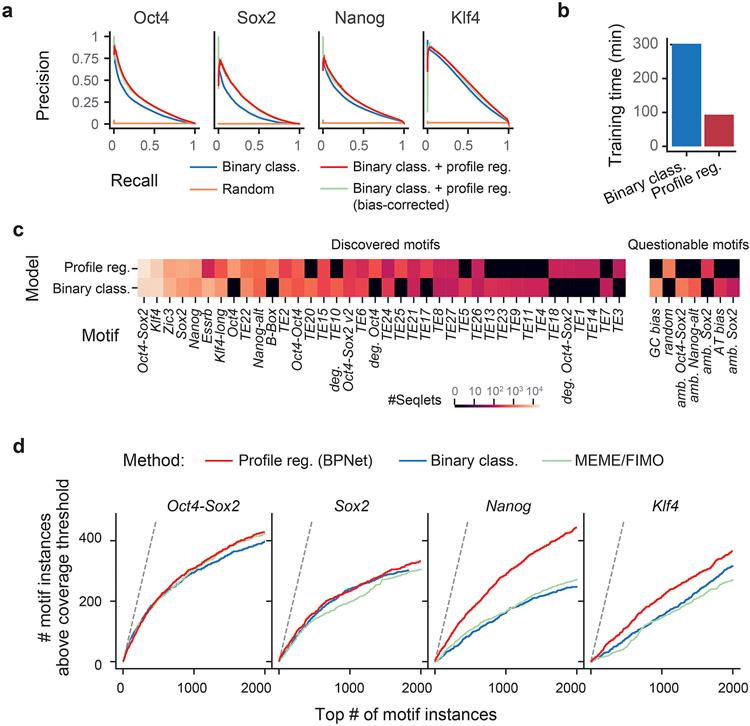
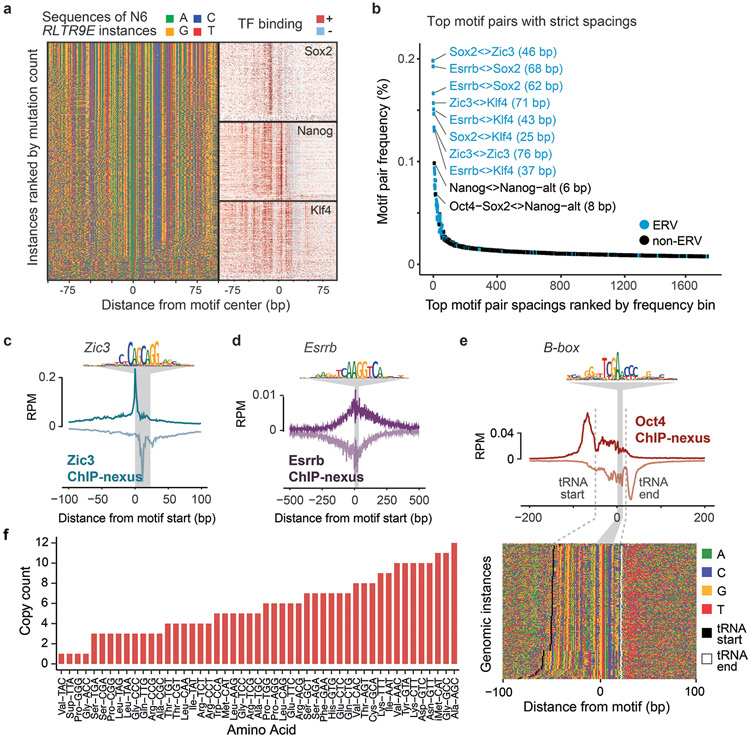
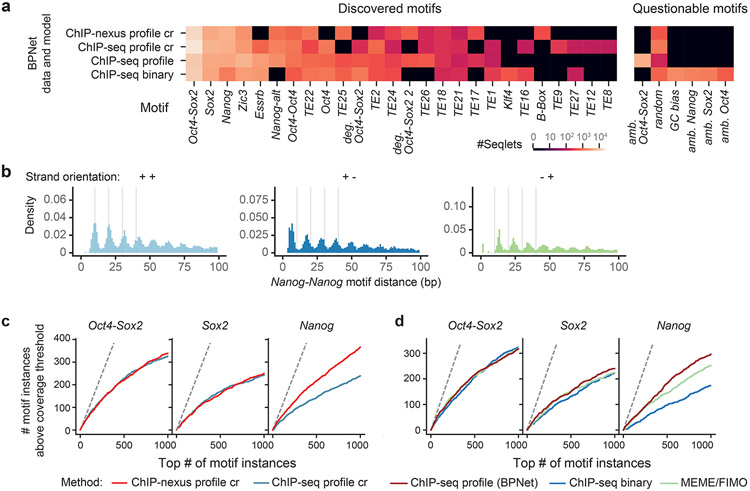
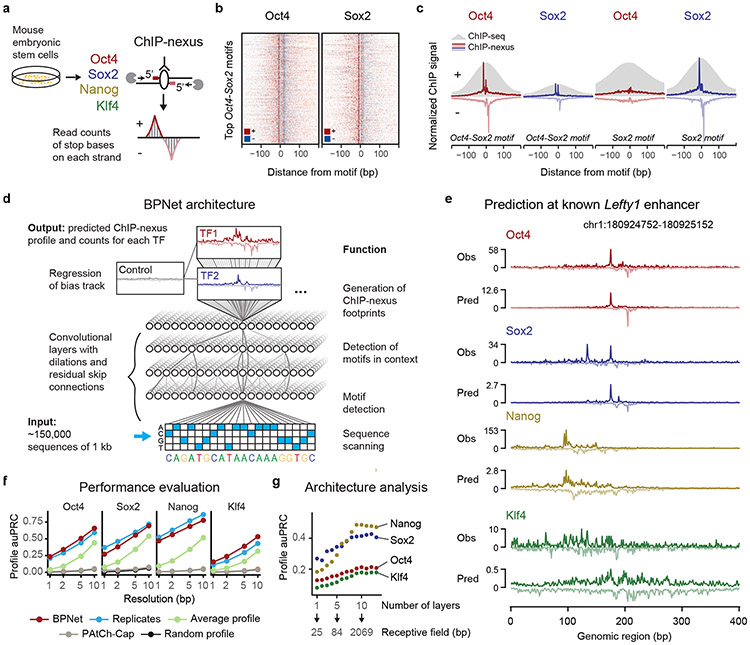
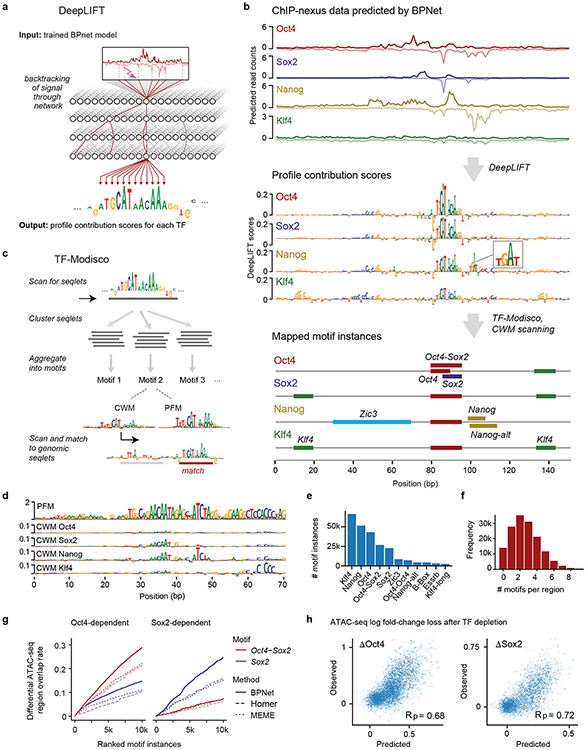
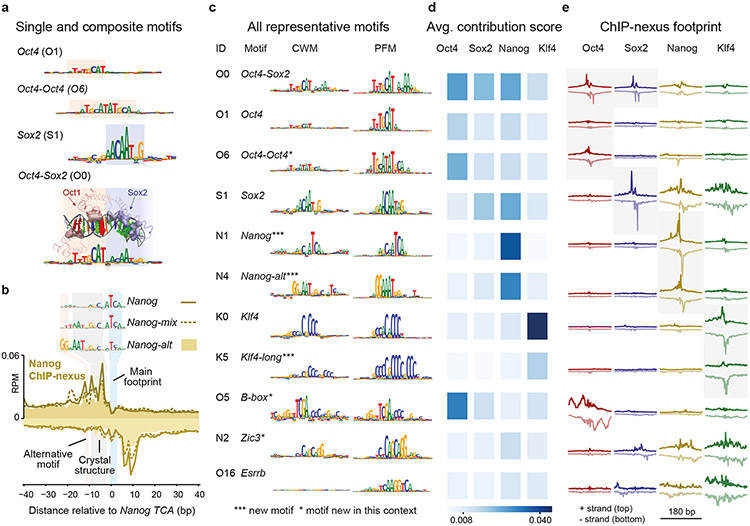
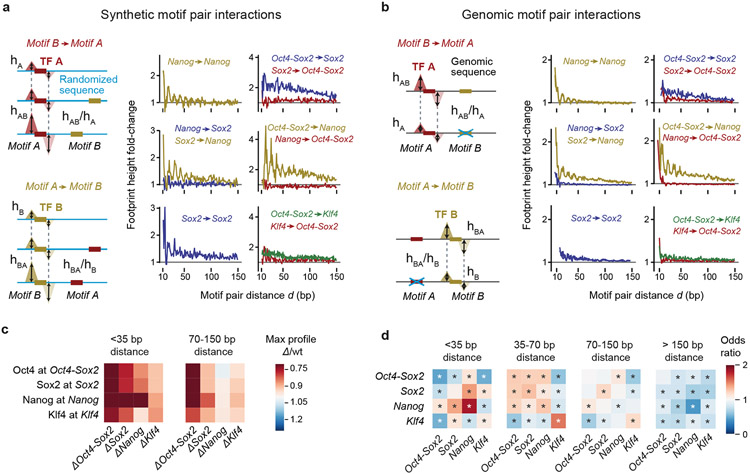
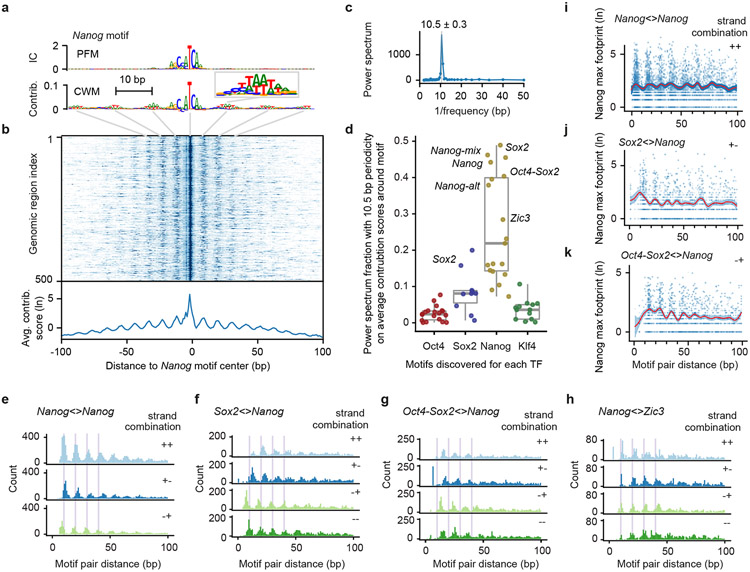
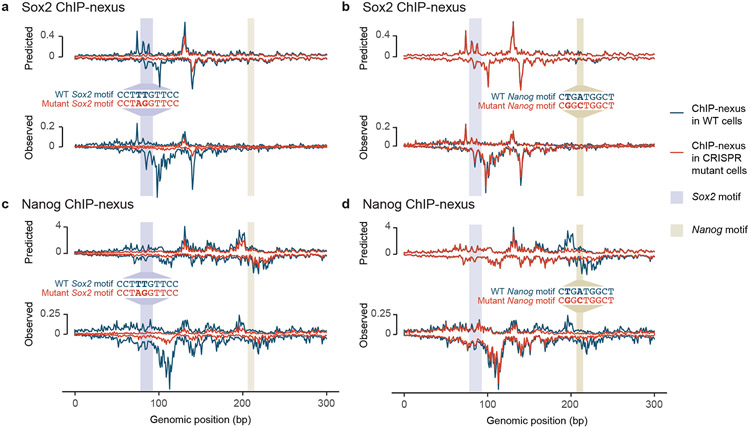
Comment in
-
Deciphering cis-regulatory grammar with deep learning.Nat Genet. 2021 Mar;53(3):266-268. doi: 10.1038/s41588-021-00814-1. Nat Genet. 2021. PMID: 33686263 No abstract available.
References
-
- Morgunova E & Taipale J Structural perspective of cooperative transcription factor binding. Curr. Opin. Struct. Biol 47, 1–8 (2017). - PubMed
-
- Zinzen RP, Senger K, Levine M & Papatsenko D Computational models for neurogenic gene expression in the Drosophila embryo. Curr. Biol 16, 1358–1365 (2006). - PubMed
Online methods references
-
- Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet j. 17, 10 (2011).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
